

A predictable conserved DNA base composition signature defines human core DNA replication origins
- PMID: 32958757
- PMCID: PMC7506530
- DOI: 10.1038/s41467-020-18527-0
A predictable conserved DNA base composition signature defines human core DNA replication origins
Abstract
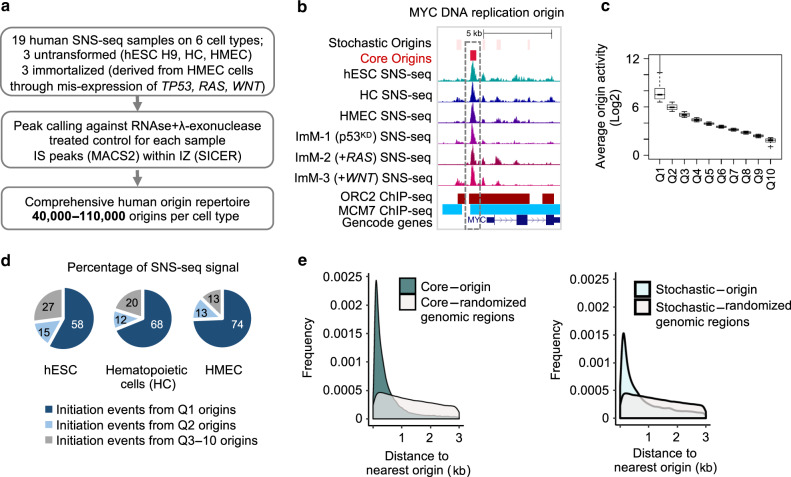
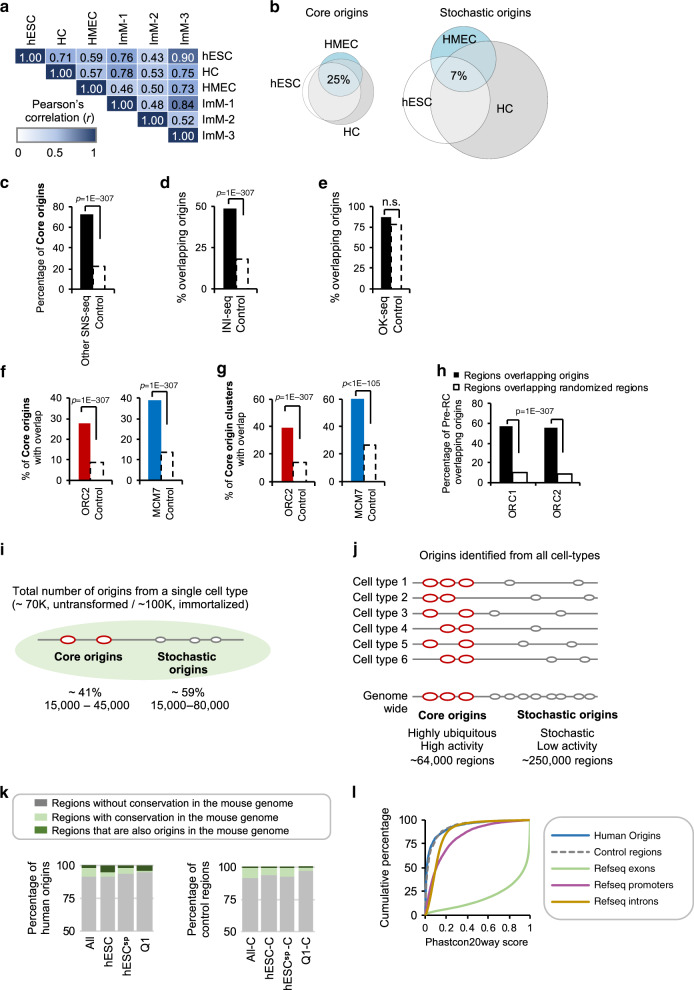
DNA replication initiates from multiple genomic locations called replication origins. In metazoa, DNA sequence elements involved in origin specification remain elusive. Here, we examine pluripotent, primary, differentiating, and immortalized human cells, and demonstrate that a class of origins, termed core origins, is shared by different cell types and host ~80% of all DNA replication initiation events in any cell population. We detect a shared G-rich DNA sequence signature that coincides with most core origins in both human and mouse genomes. Transcription and G-rich elements can independently associate with replication origin activity. Computational algorithms show that core origins can be predicted, based solely on DNA sequence patterns but not on consensus motifs. Our results demonstrate that, despite an attributed stochasticity, core origins are chosen from a limited pool of genomic regions. Immortalization through oncogenic gene expression, but not normal cellular differentiation, results in increased stochastic firing from heterochromatin and decreased origin density at TAD borders.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
