

Reconstructing organisms in silico: genome-scale models and their emerging applications
- PMID: 32958892
- PMCID: PMC7981288
- DOI: 10.1038/s41579-020-00440-4
Reconstructing organisms in silico: genome-scale models and their emerging applications
Abstract
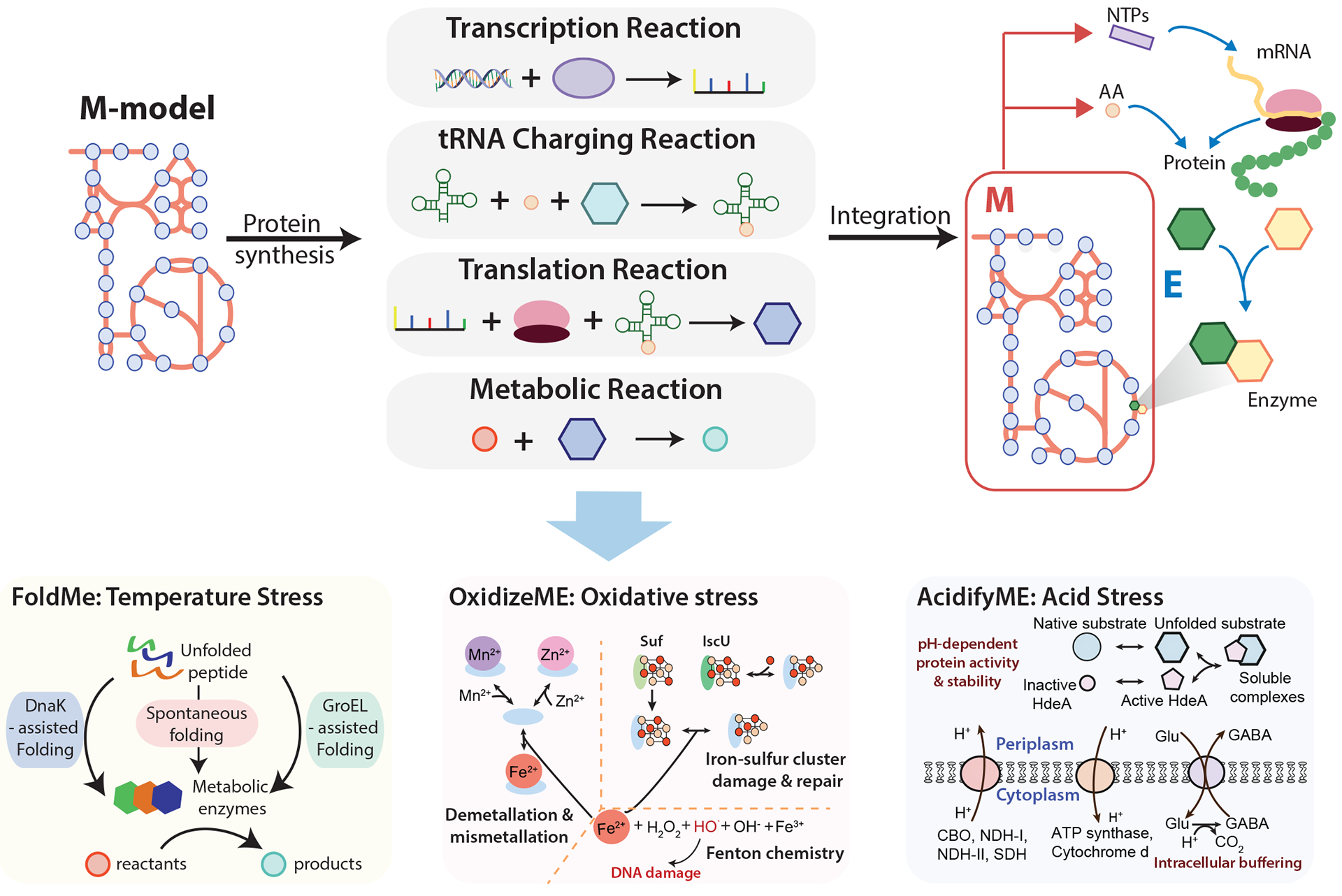
Escherichia coli is considered to be the best-known microorganism given the large number of published studies detailing its genes, its genome and the biochemical functions of its molecular components. This vast literature has been systematically assembled into a reconstruction of the biochemical reaction networks that underlie E. coli's functions, a process which is now being applied to an increasing number of microorganisms. Genome-scale reconstructed networks are organized and systematized knowledge bases that have multiple uses, including conversion into computational models that interpret and predict phenotypic states and the consequences of environmental and genetic perturbations. These genome-scale models (GEMs) now enable us to develop pan-genome analyses that provide mechanistic insights, detail the selection pressures on proteome allocation and address stress phenotypes. In this Review, we first discuss the overall development of GEMs and their applications. Next, we review the evolution of the most complete GEM that has been developed to date: the E. coli GEM. Finally, we explore three emerging areas in genome-scale modelling of microbial phenotypes: collections of strain-specific models, metabolic and macromolecular expression models, and simulation of stress responses.
© 2020. Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no conflict of interest
Figures
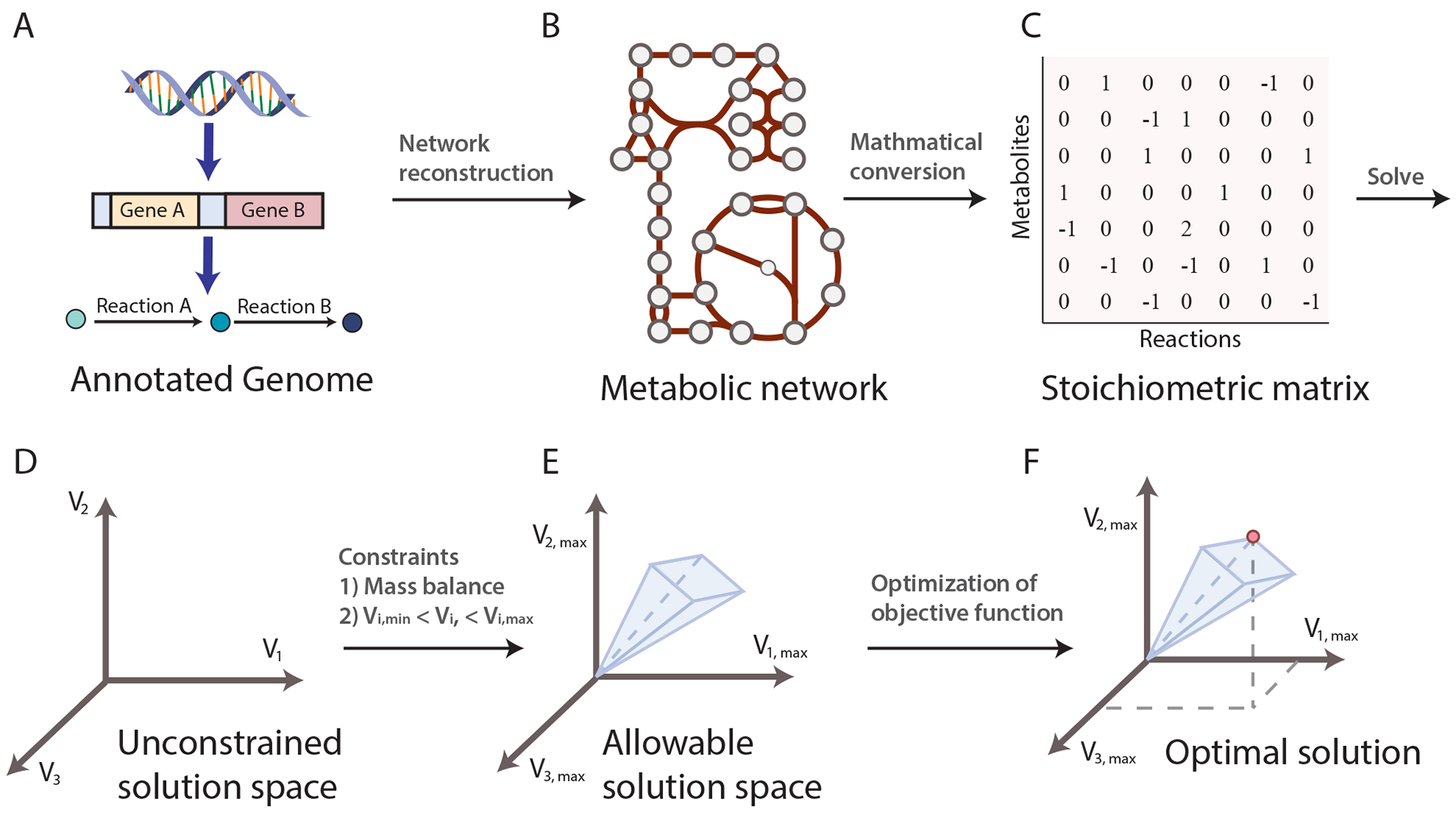
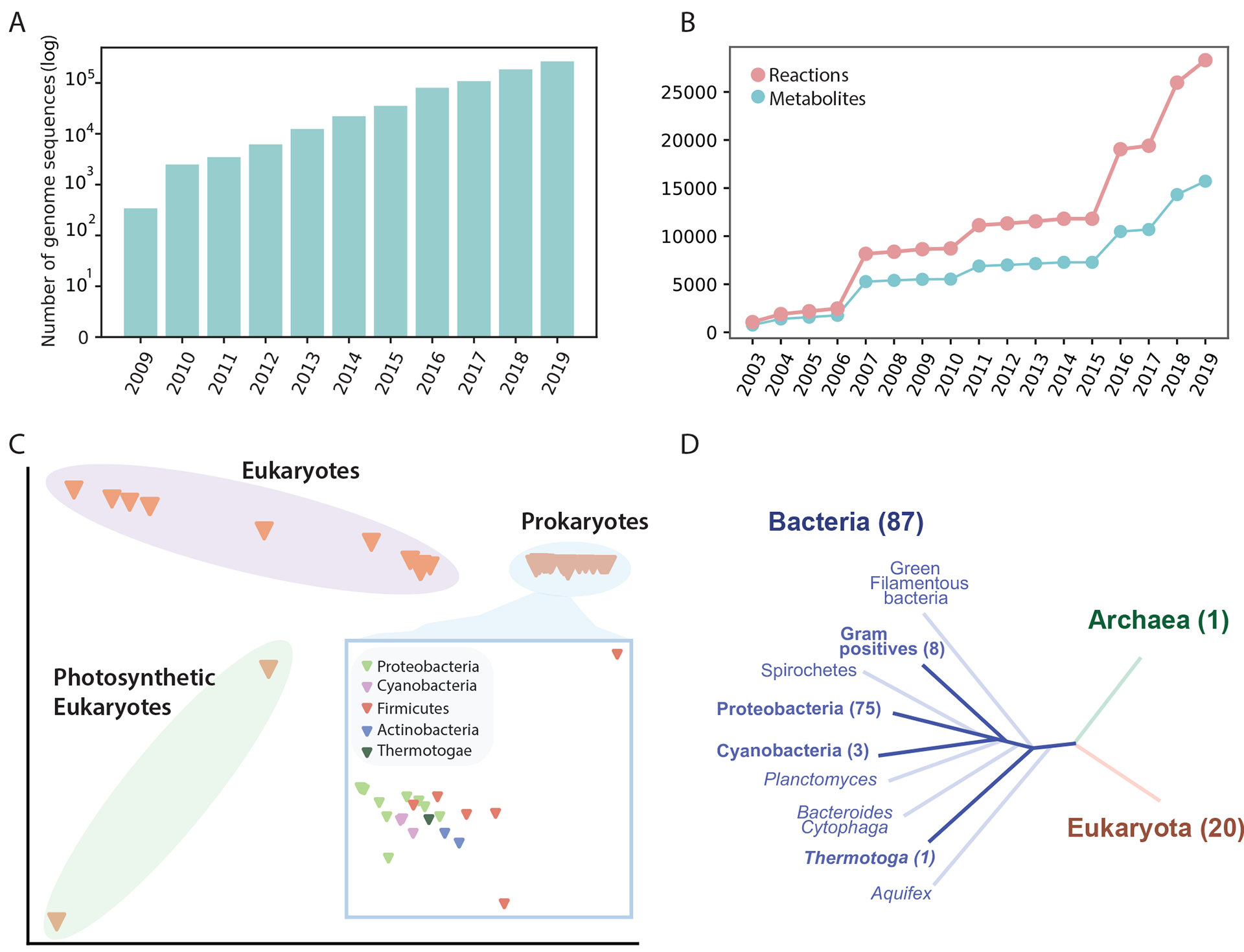
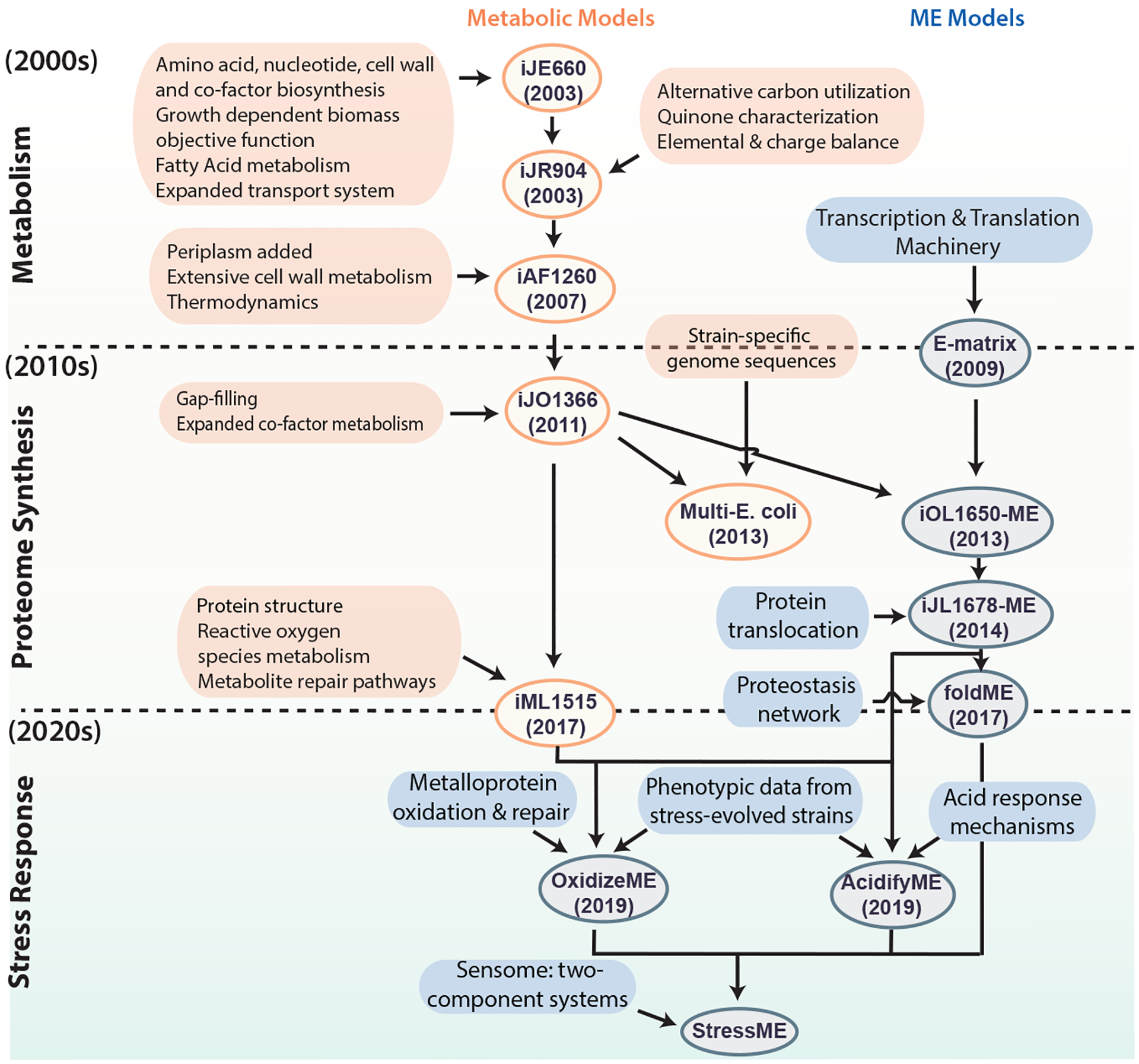
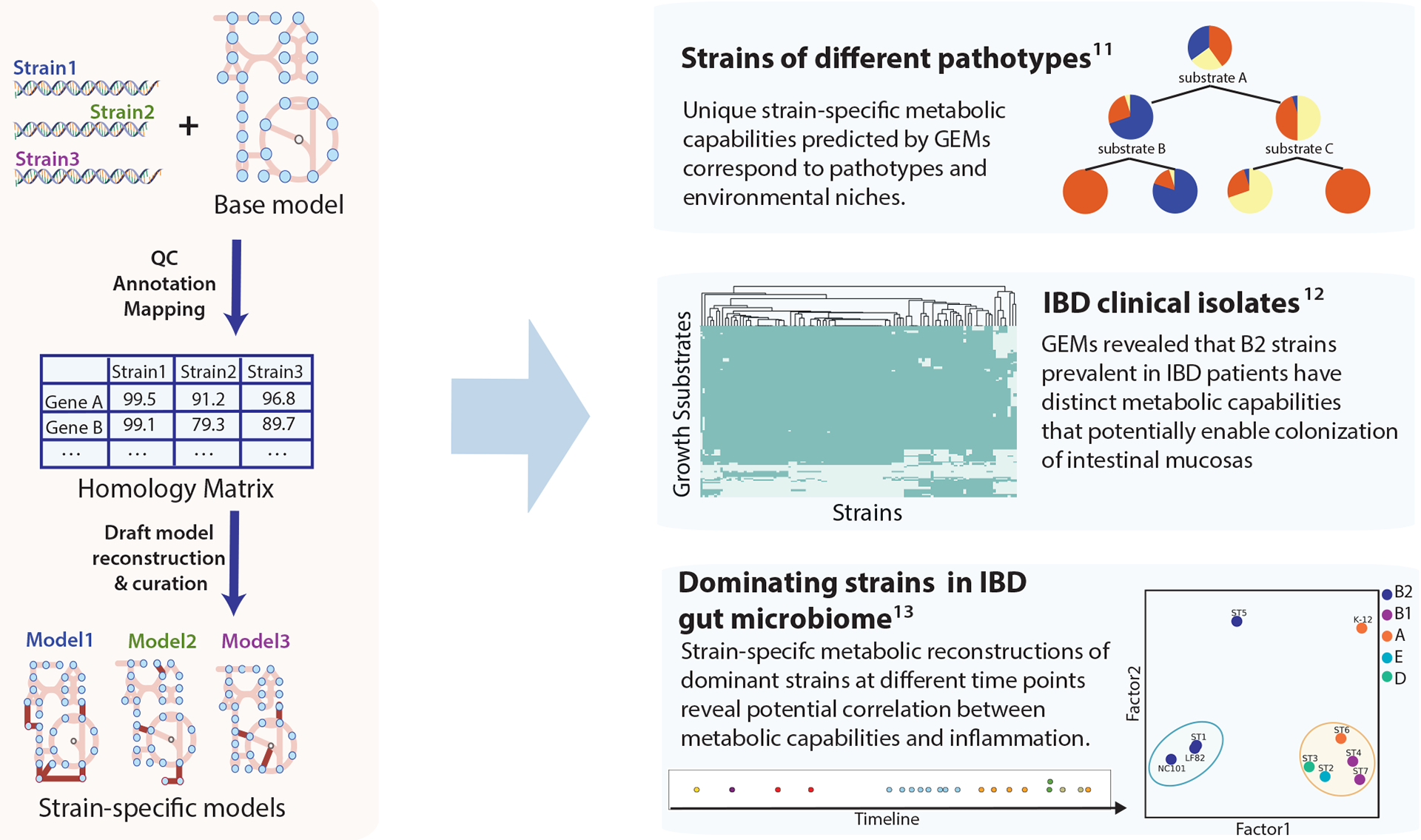
References
-
- Price ND, Reed JL & Palsson BØ Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol 2, 886–897 (2004). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
