

Transcription imparts architecture, function and logic to enhancer units
- PMID: 32958950
- PMCID: PMC7541647
- DOI: 10.1038/s41588-020-0686-2
Transcription imparts architecture, function and logic to enhancer units
Abstract
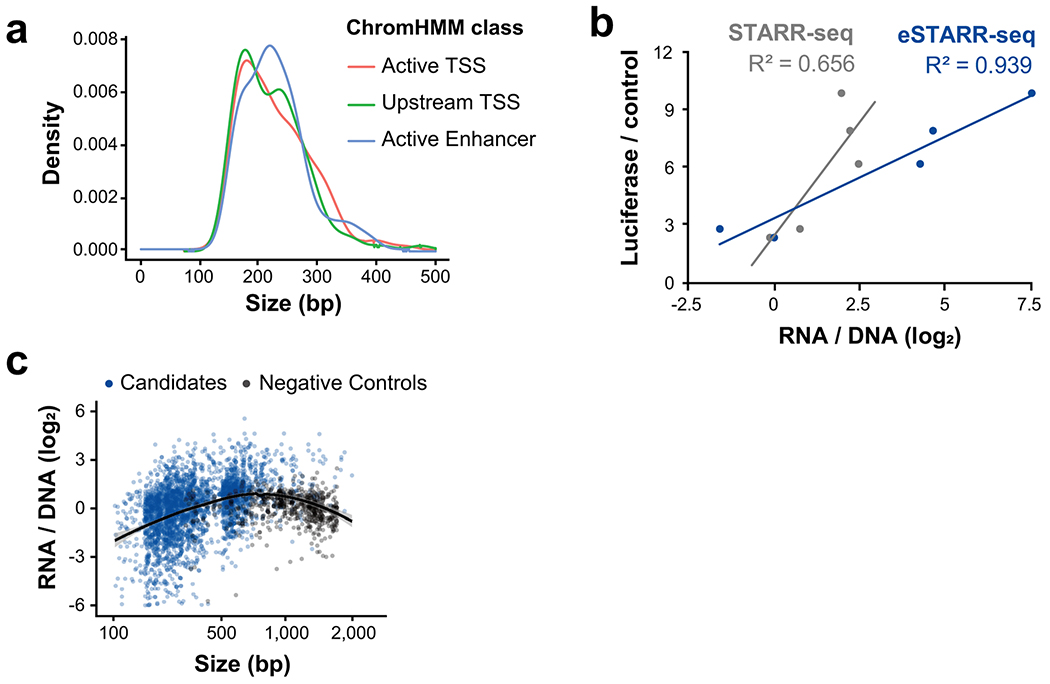
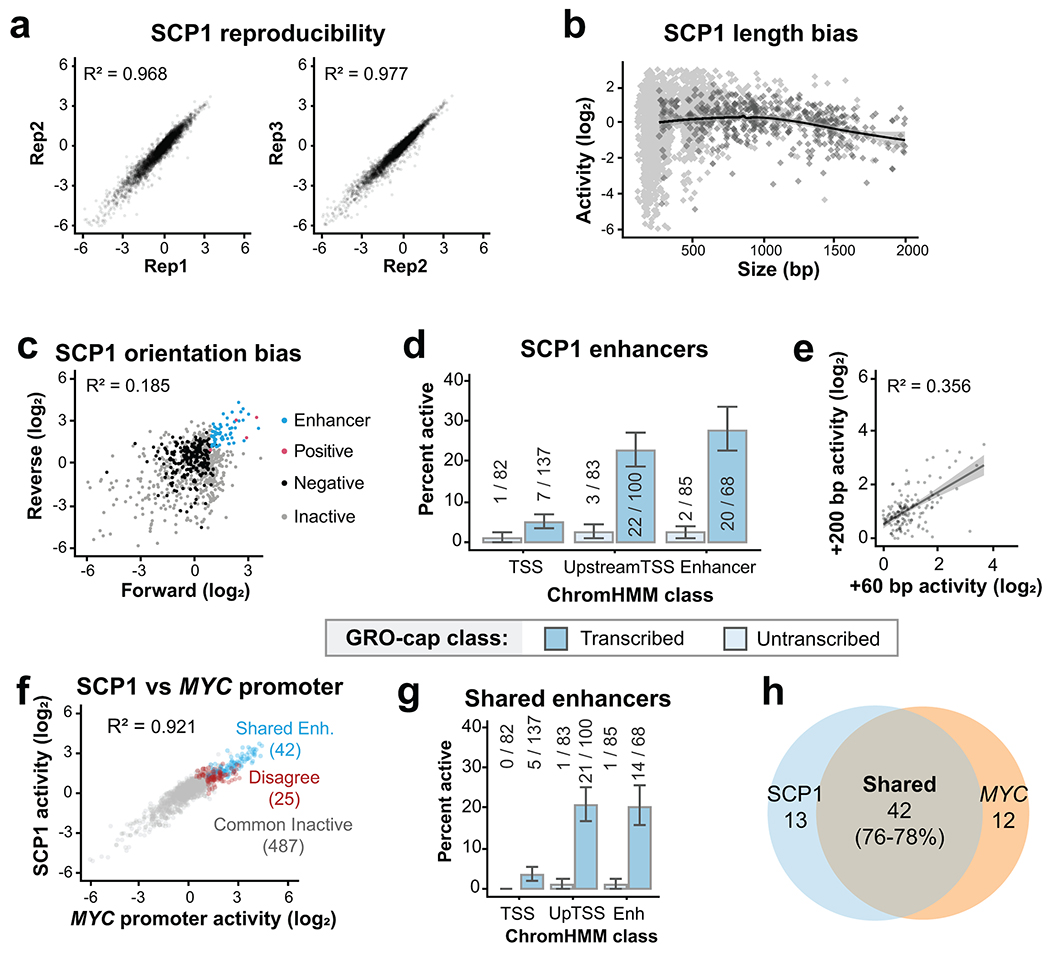
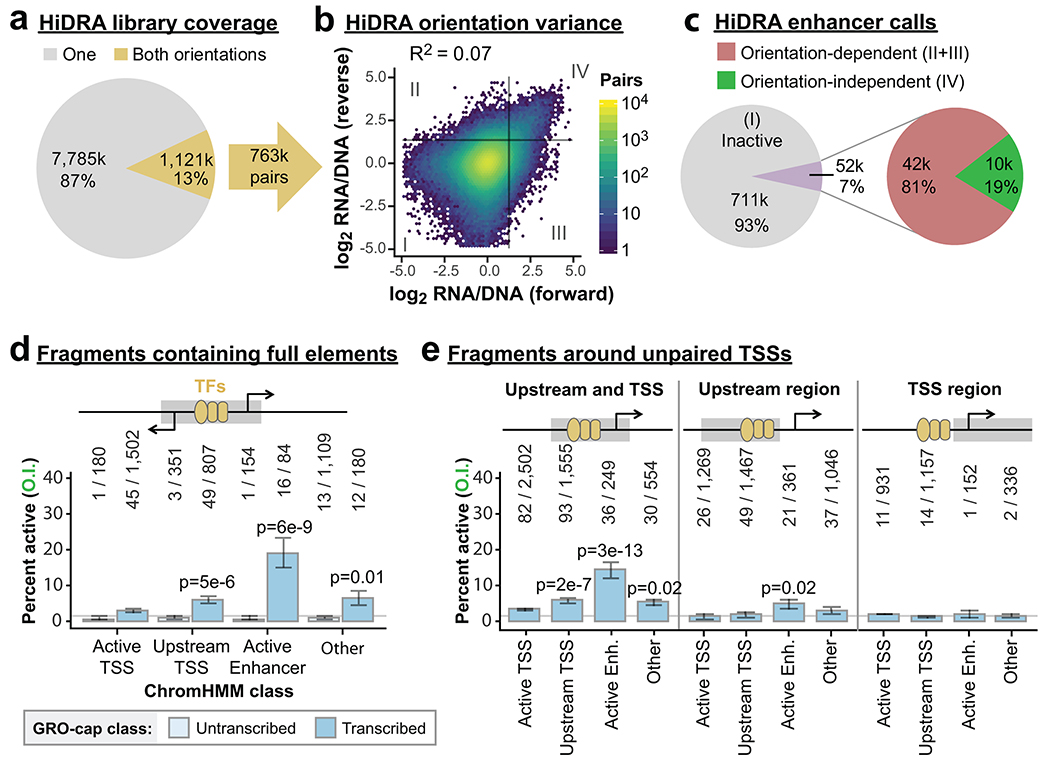
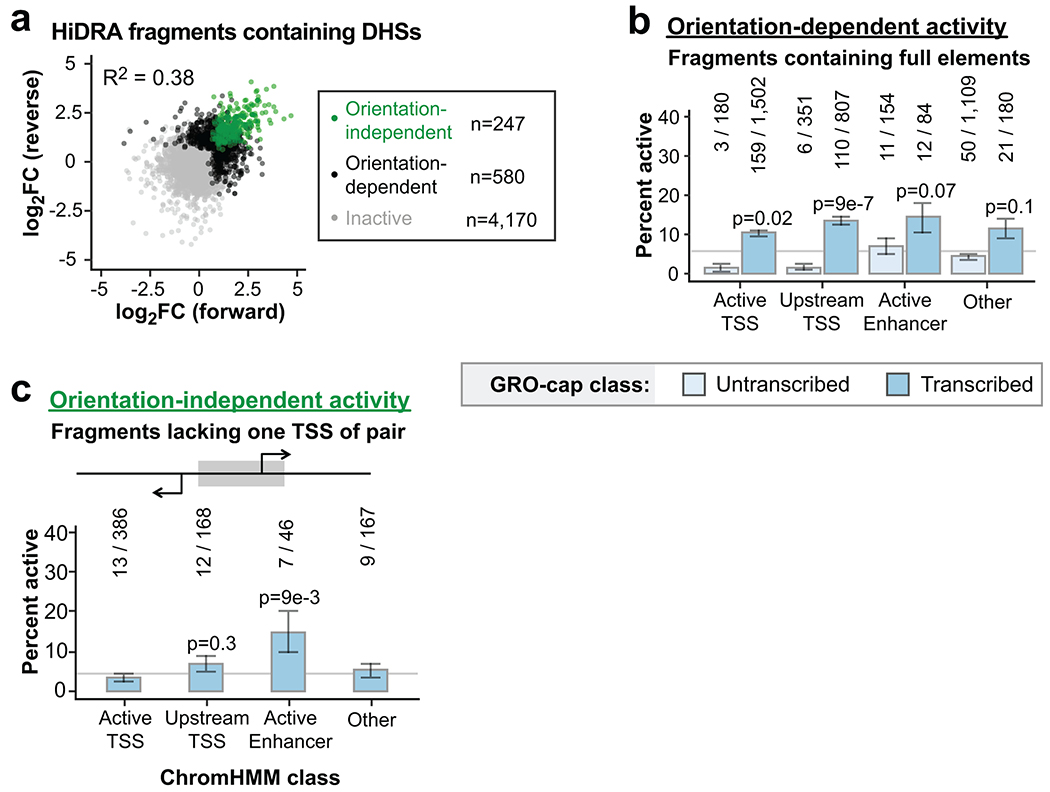
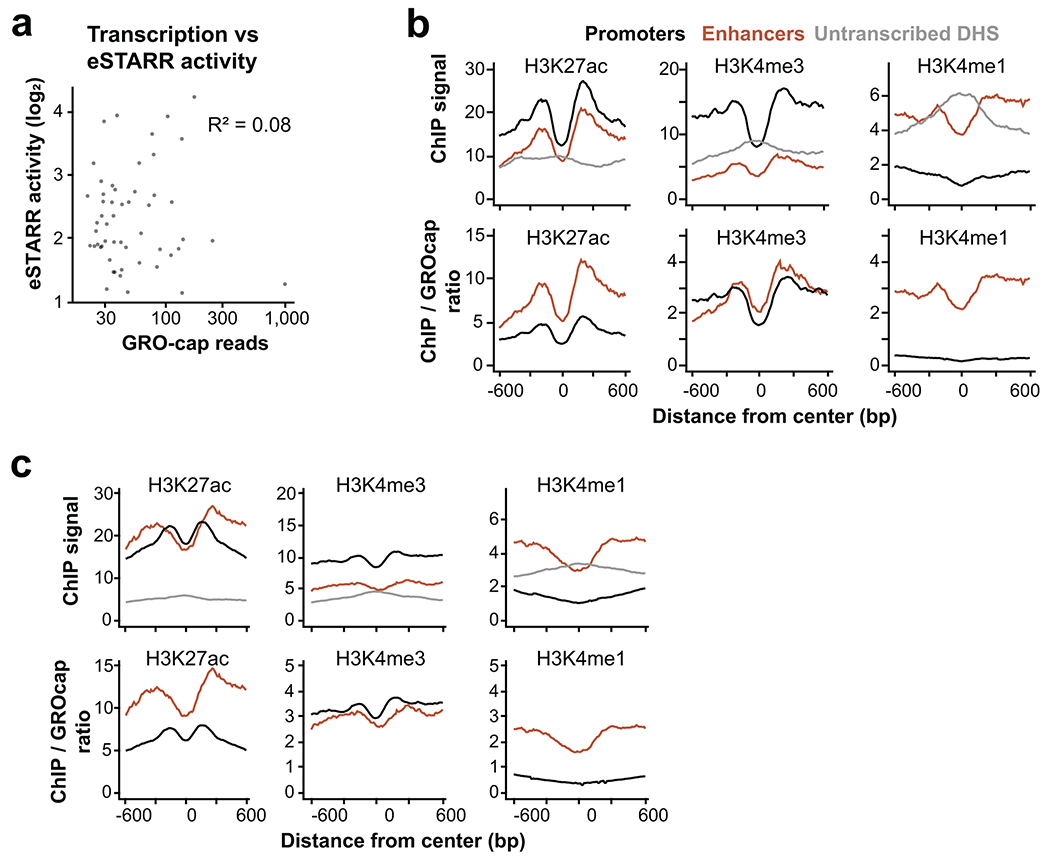
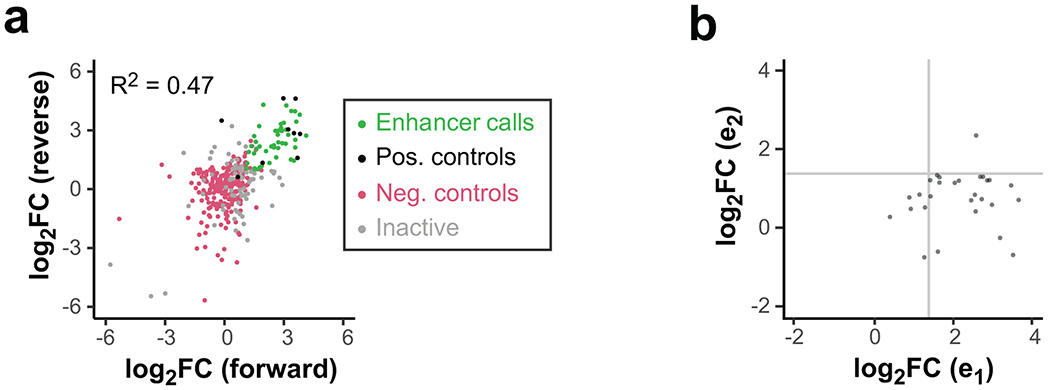
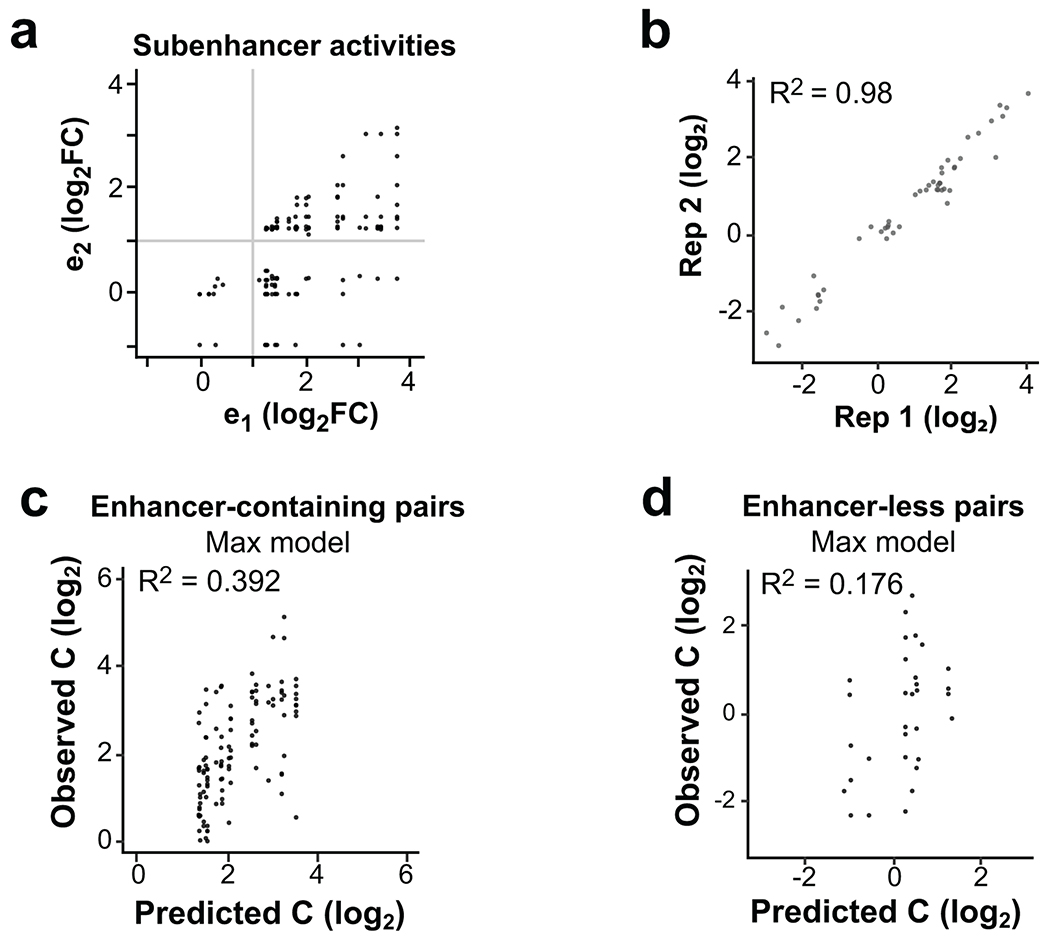
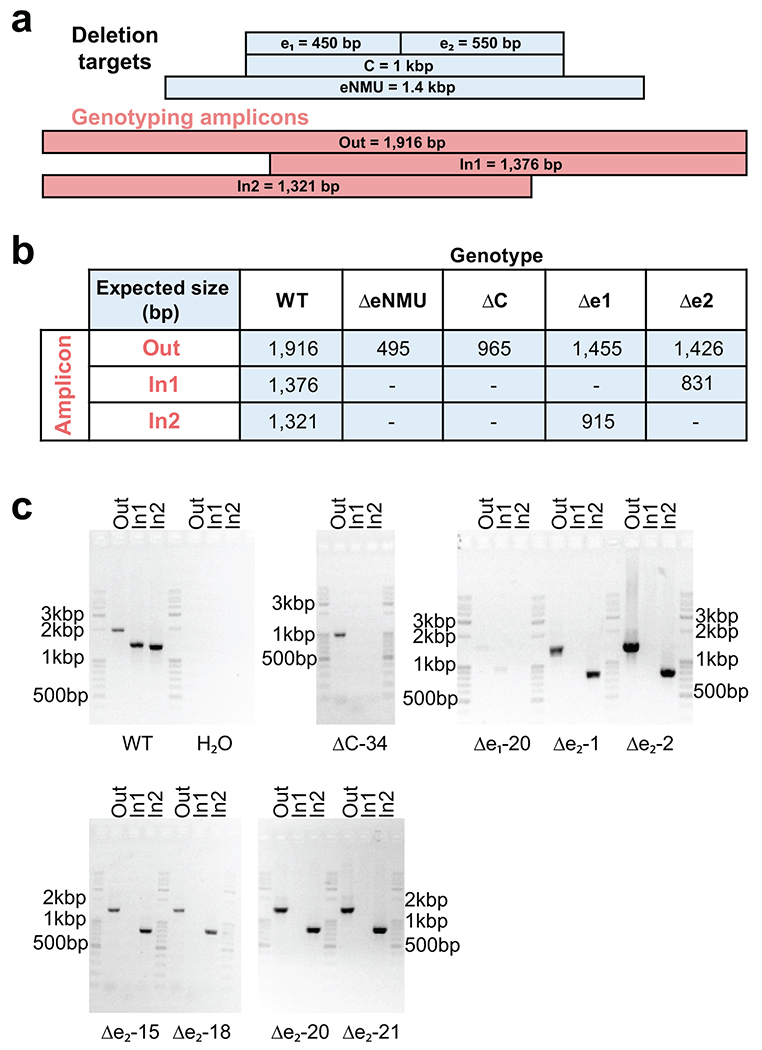
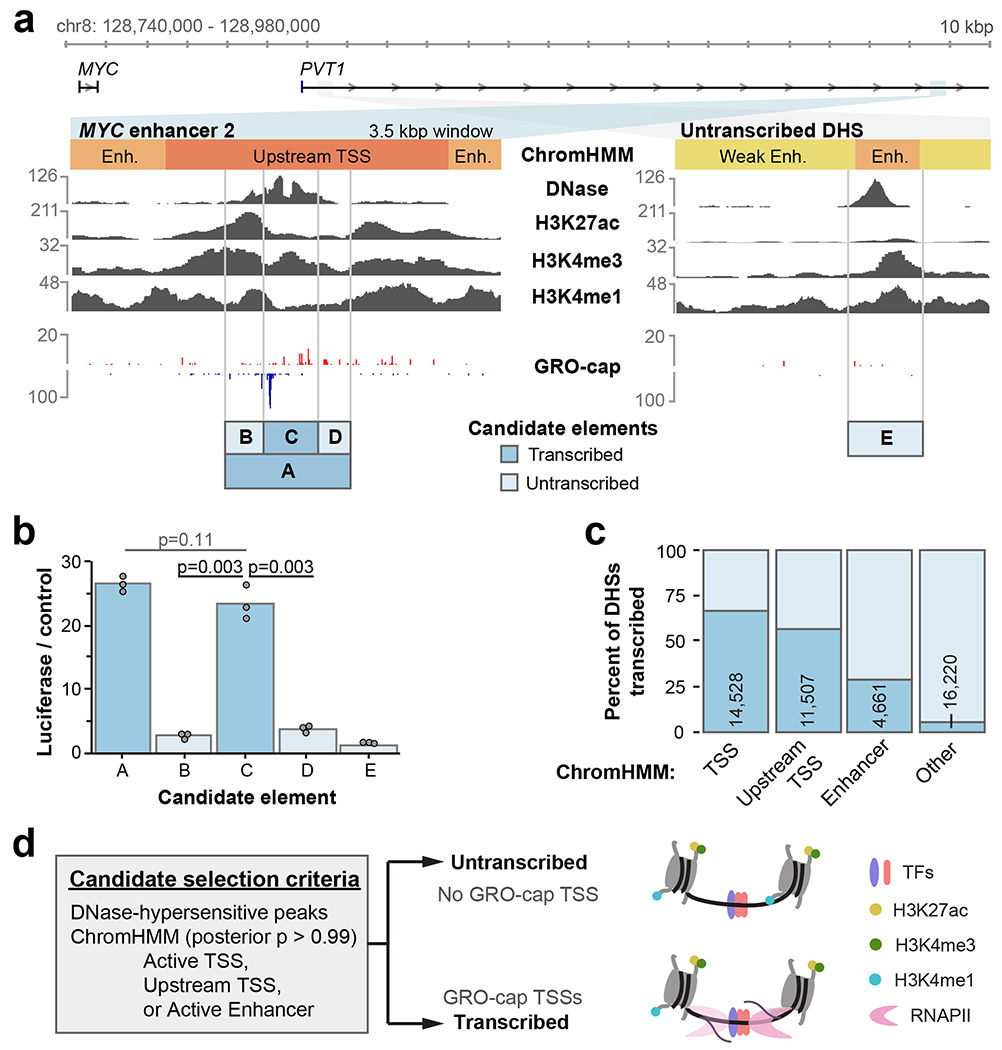
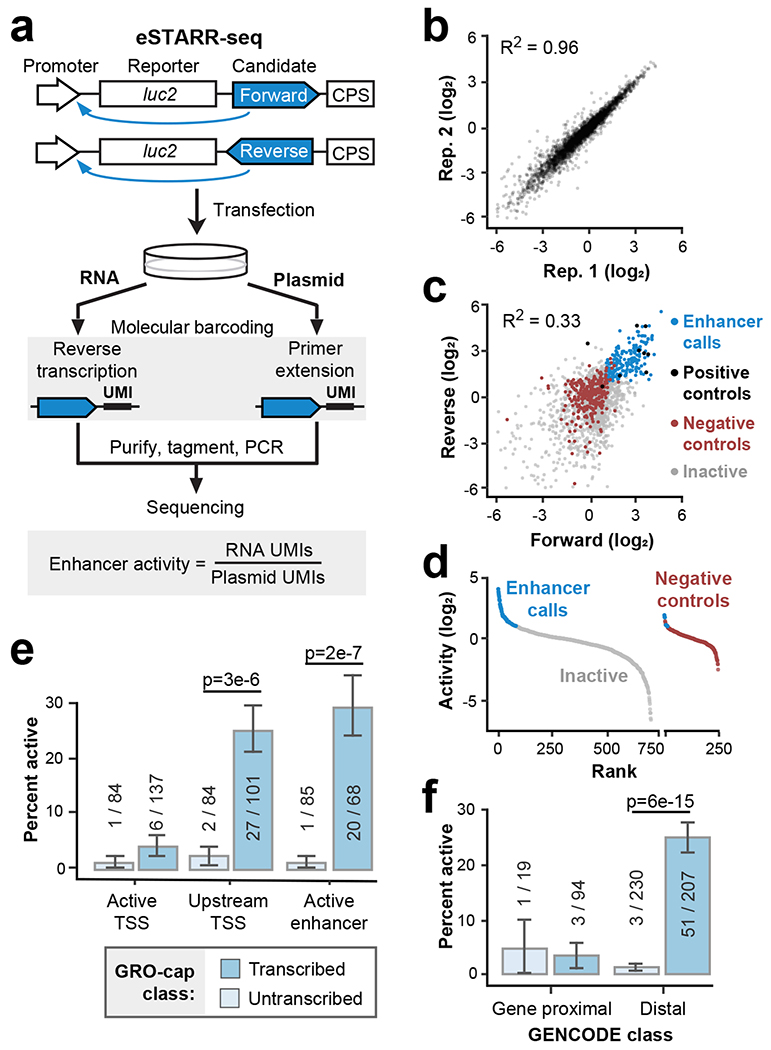
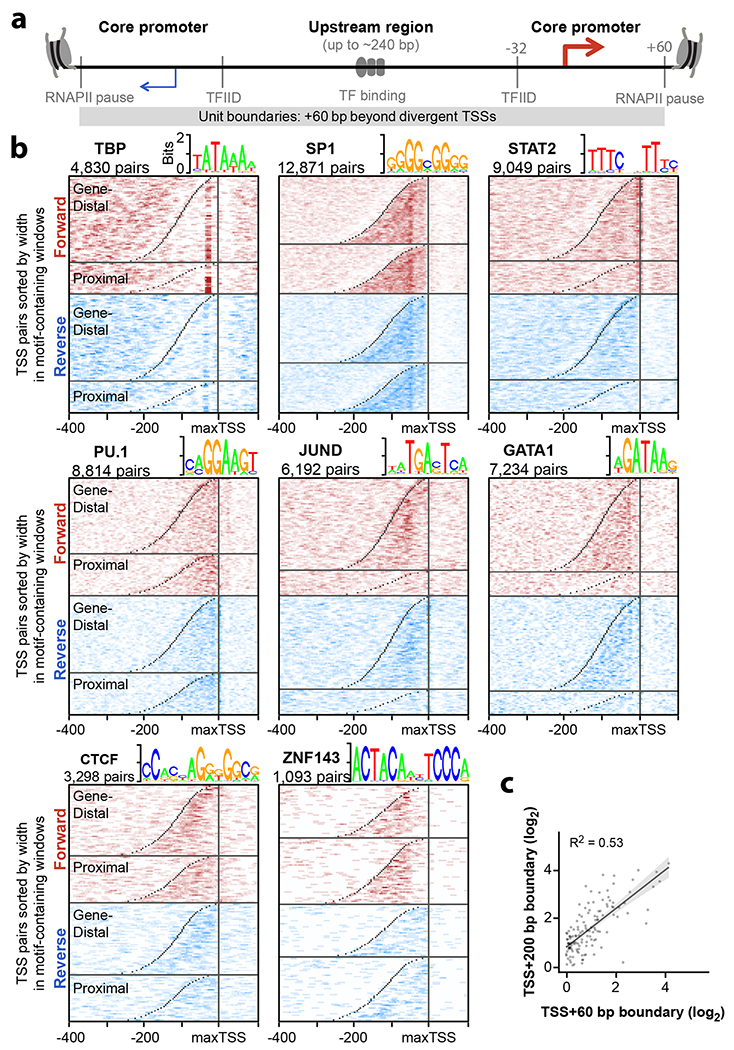
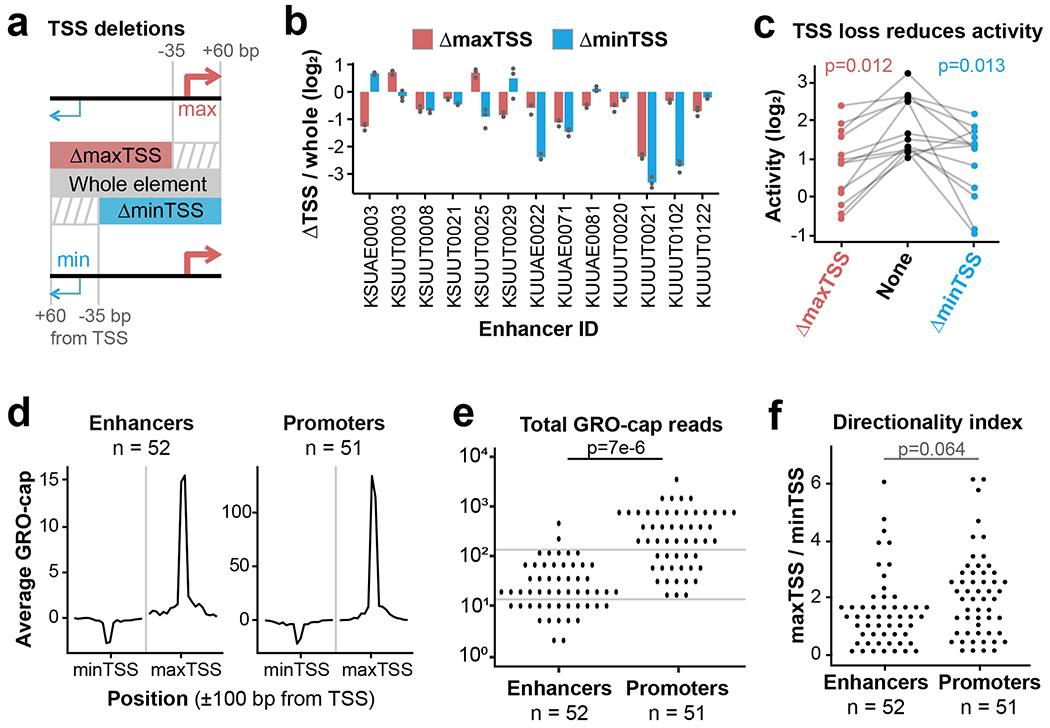
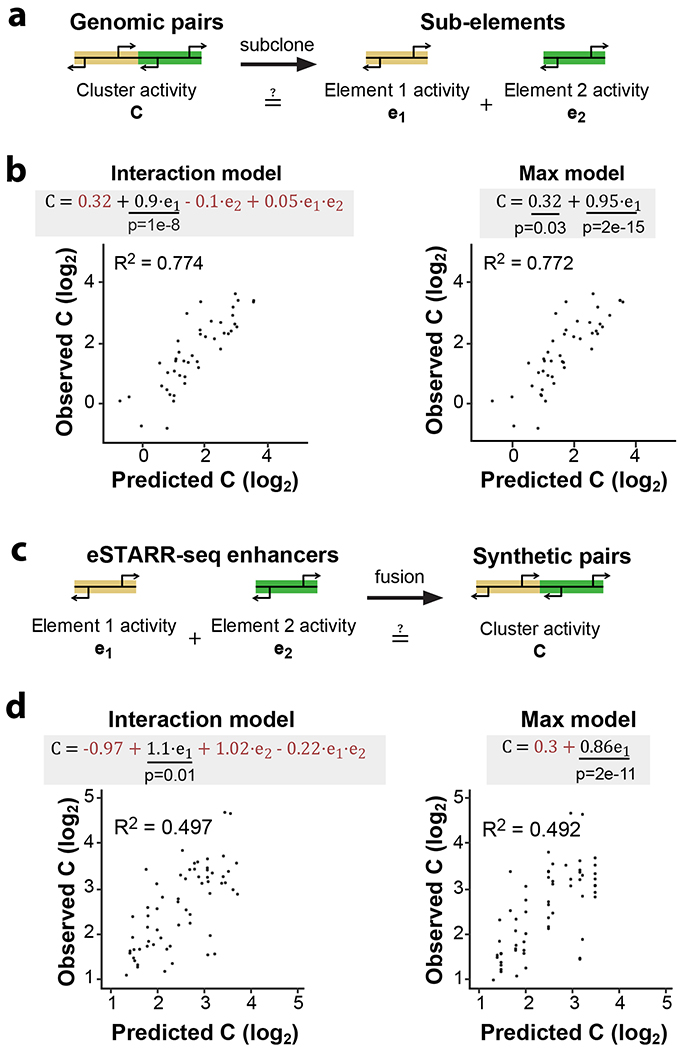
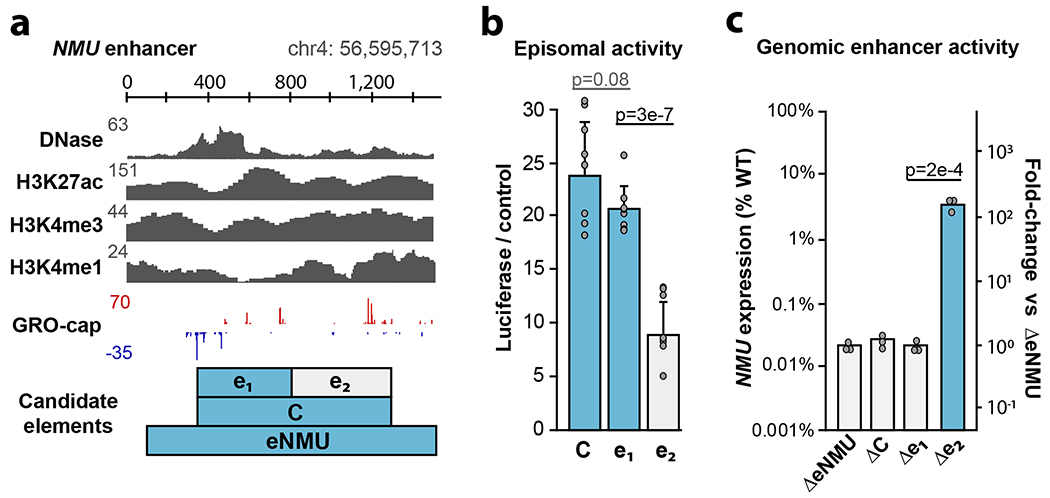
Distal enhancers play pivotal roles in development and disease yet remain one of the least understood regulatory elements. We used massively parallel reporter assays to perform functional comparisons of two leading enhancer models and find that gene-distal transcription start sites are robust predictors of active enhancers with higher resolution than histone modifications. We show that active enhancer units are precisely delineated by active transcription start sites, validate that these boundaries are sufficient for capturing enhancer function, and confirm that core promoter sequences are necessary for this activity. We assay adjacent enhancers and find that their joint activity is often driven by the stronger unit within the cluster. Finally, we validate these results through functional dissection of a distal enhancer cluster using CRISPR-Cas9 deletions. In summary, definition of high-resolution enhancer boundaries enables deconvolution of complex regulatory loci into modular units.
Conflict of interest statement
Competing interests
None.
Figures
References
-
- Serfling E, Jasin M & Schaffner W Enhancers and eukaryotic gene transcription. Trends in Genetics 1, 224–230 (1985).
-
- Arnold CD et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–7 (2013). - PubMed
-
- Orkin SH Regulation of globin gene expression in erythroid cells. Eur J Biochem 231, 271–81 (1995). - PubMed
Methods-only references
-
- Arad U Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. Biotechniques 24, 760–2 (1998). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
