

Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency
- PMID: 32960813
- PMCID: PMC7773360
- DOI: 10.1172/JCI139980
Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency
Abstract
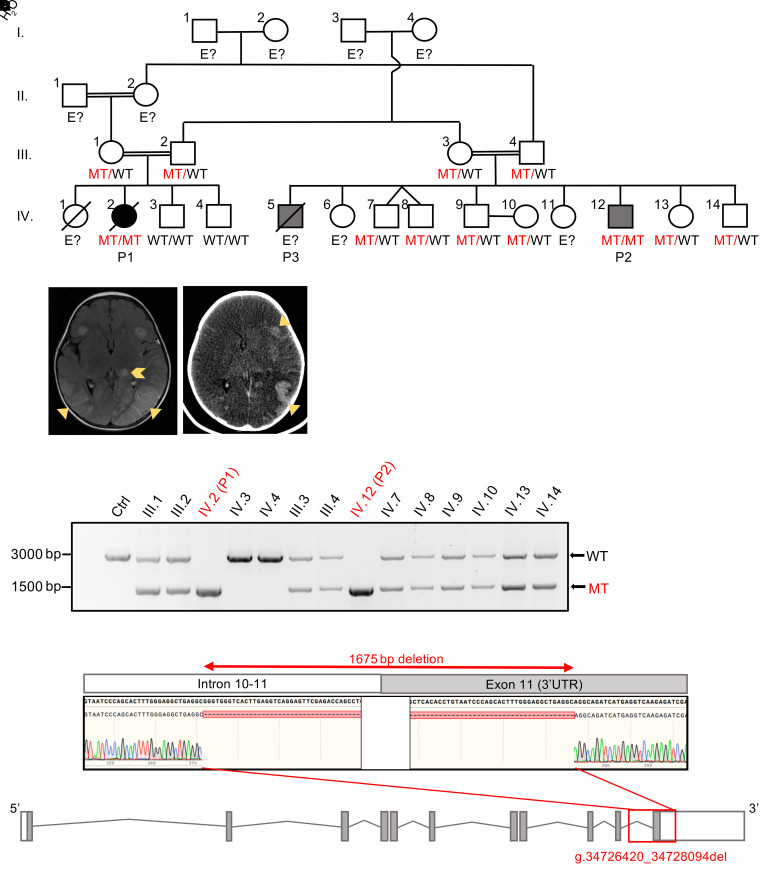
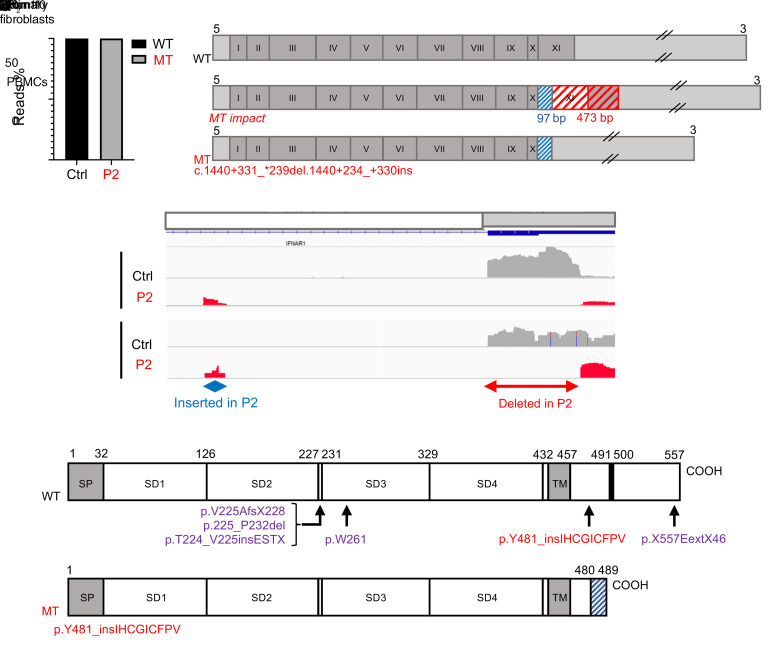
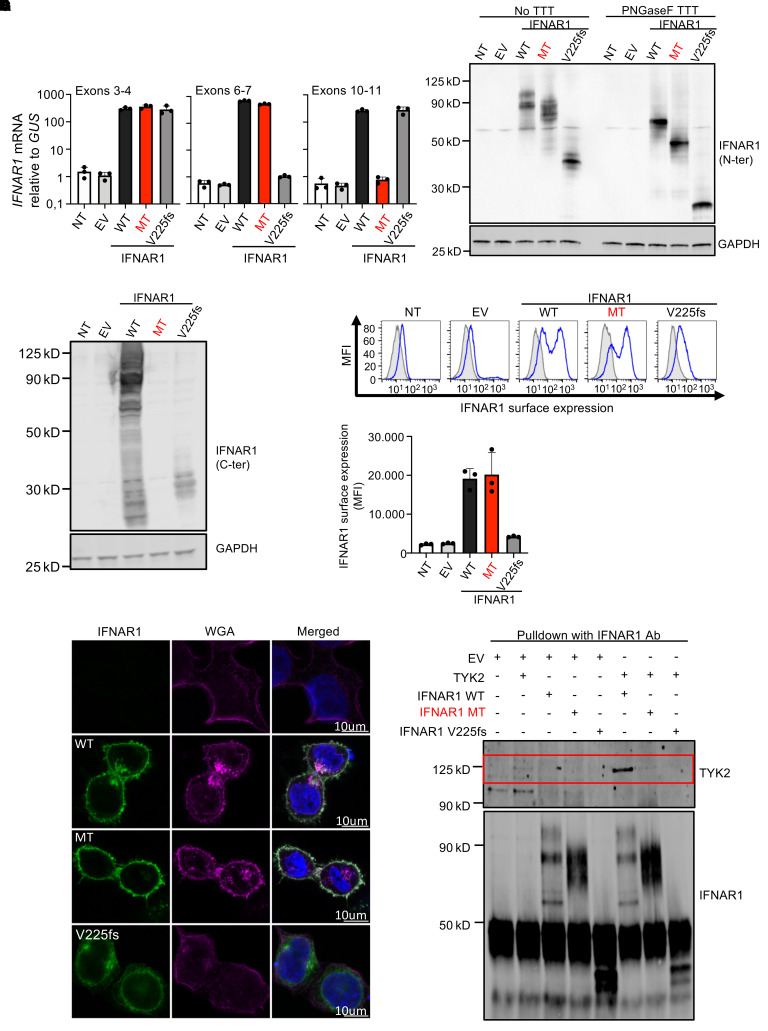
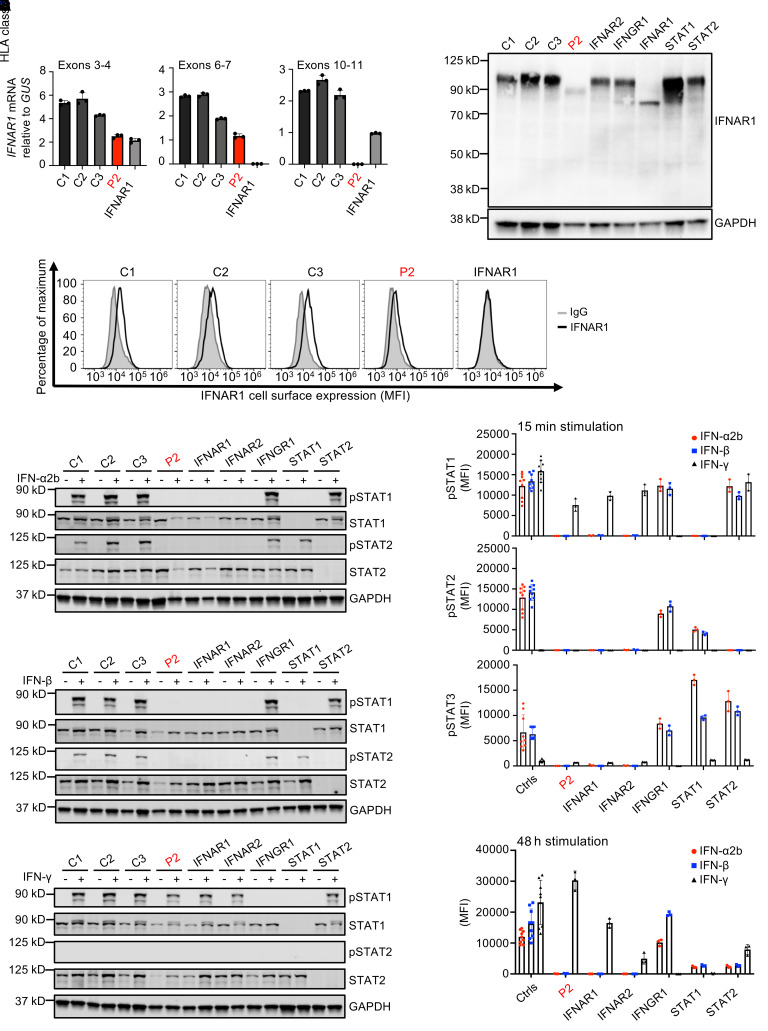
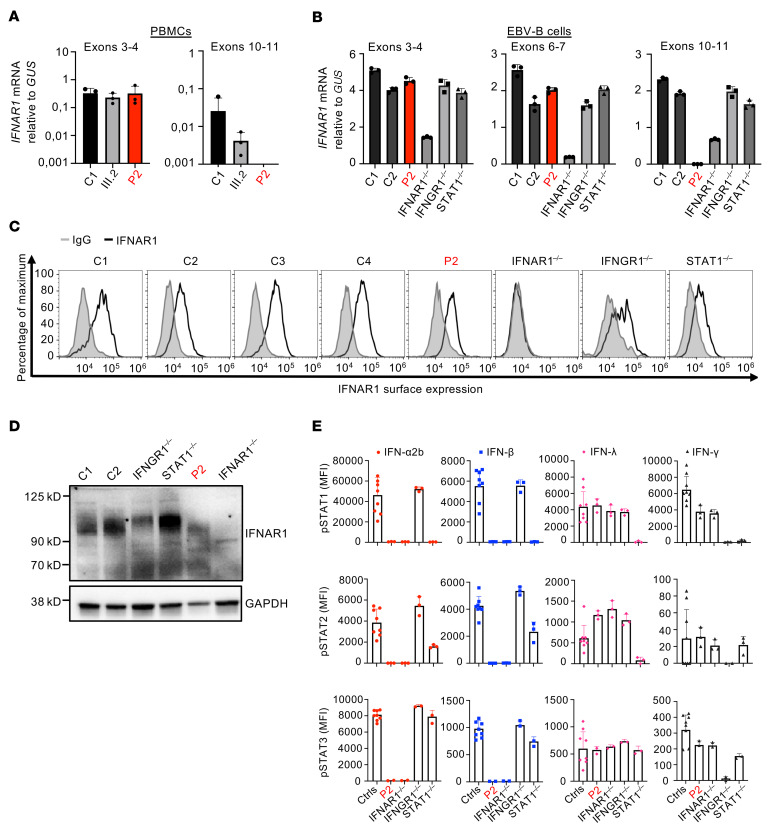
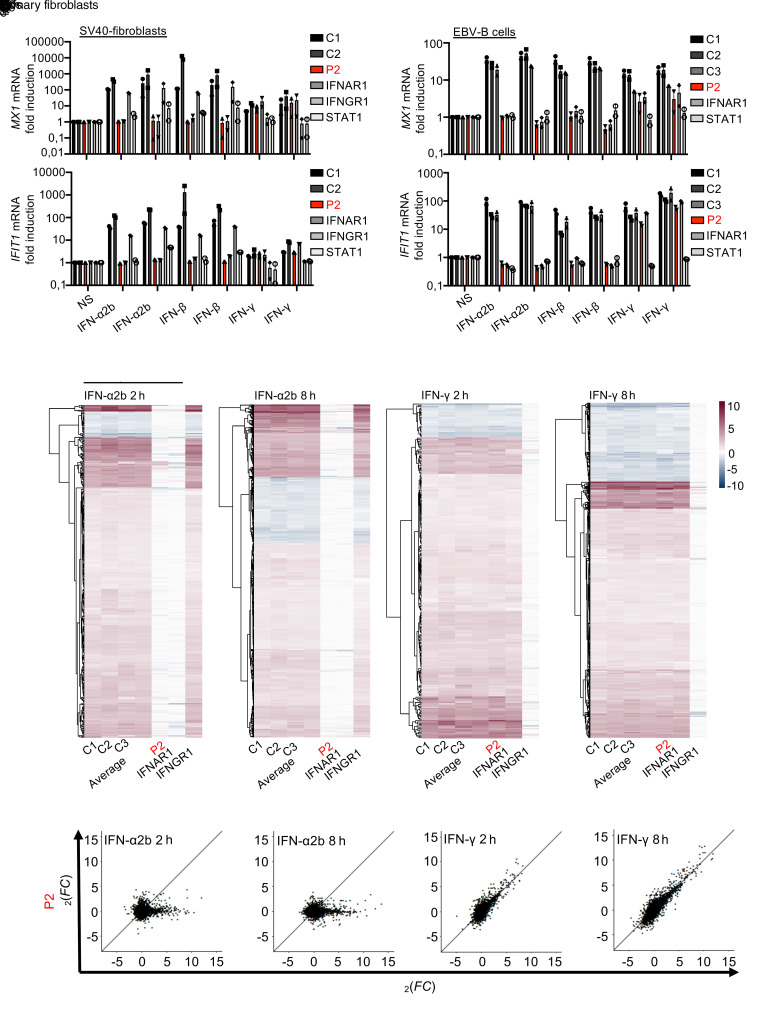
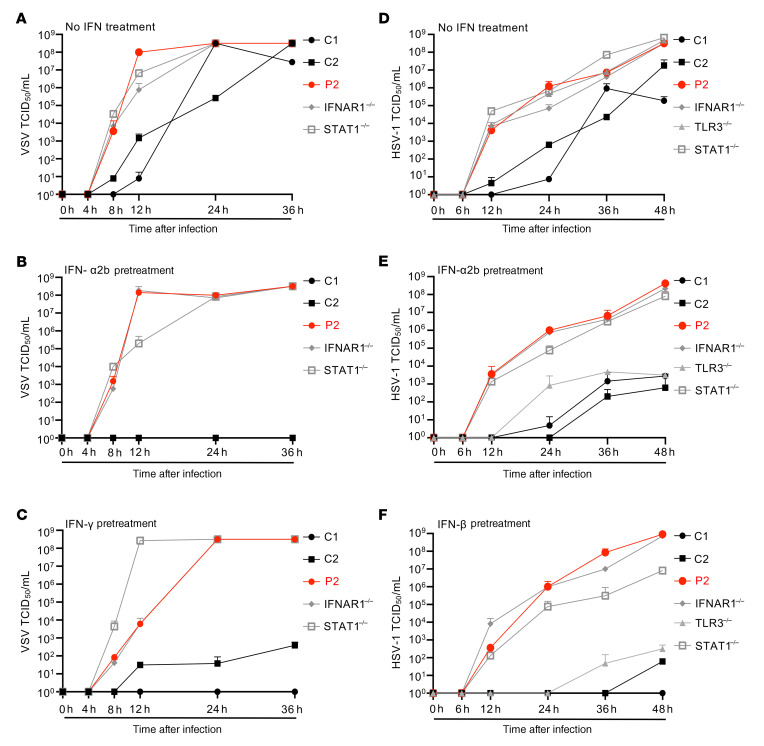
Inborn errors of TLR3-dependent IFN-α/β- and IFN-λ-mediated immunity in the CNS can underlie herpes simplex virus 1 (HSV-1) encephalitis (HSE). The respective contributions of IFN-α/β and IFN-λ are unknown. We report a child homozygous for a genomic deletion of the entire coding sequence and part of the 3'-UTR of the last exon of IFNAR1, who died of HSE at the age of 2 years. An older cousin died following vaccination against measles, mumps, and rubella at 12 months of age, and another 17-year-old cousin homozygous for the same variant has had other, less severe, viral illnesses. The encoded IFNAR1 protein is expressed on the cell surface but is truncated and cannot interact with the tyrosine kinase TYK2. The patient's fibroblasts and EBV-B cells did not respond to IFN-α2b or IFN-β, in terms of STAT1, STAT2, and STAT3 phosphorylation or the genome-wide induction of IFN-stimulated genes. The patient's fibroblasts were susceptible to viruses, including HSV-1, even in the presence of exogenous IFN-α2b or IFN-β. HSE is therefore a consequence of inherited complete IFNAR1 deficiency. This viral disease occurred in natural conditions, unlike those previously reported in other patients with IFNAR1 or IFNAR2 deficiency. This experiment of nature indicates that IFN-α/β are essential for anti-HSV-1 immunity in the CNS.
Keywords: Cytokines; Genetic diseases; Immunology; Infectious disease; Innate immunity.
Conflict of interest statement
Figures
References
-
- Whitley RJ. Herpes simplex virus infections of the central nervous system. Continuum (Minneap Minn) 2015;21(6 Neuroinfectious Disease):1704–1713. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
