

Doxorubicin-induced p53 interferes with mitophagy in cardiac fibroblasts
- PMID: 32960902
- PMCID: PMC7508395
- DOI: 10.1371/journal.pone.0238856
Doxorubicin-induced p53 interferes with mitophagy in cardiac fibroblasts
Abstract
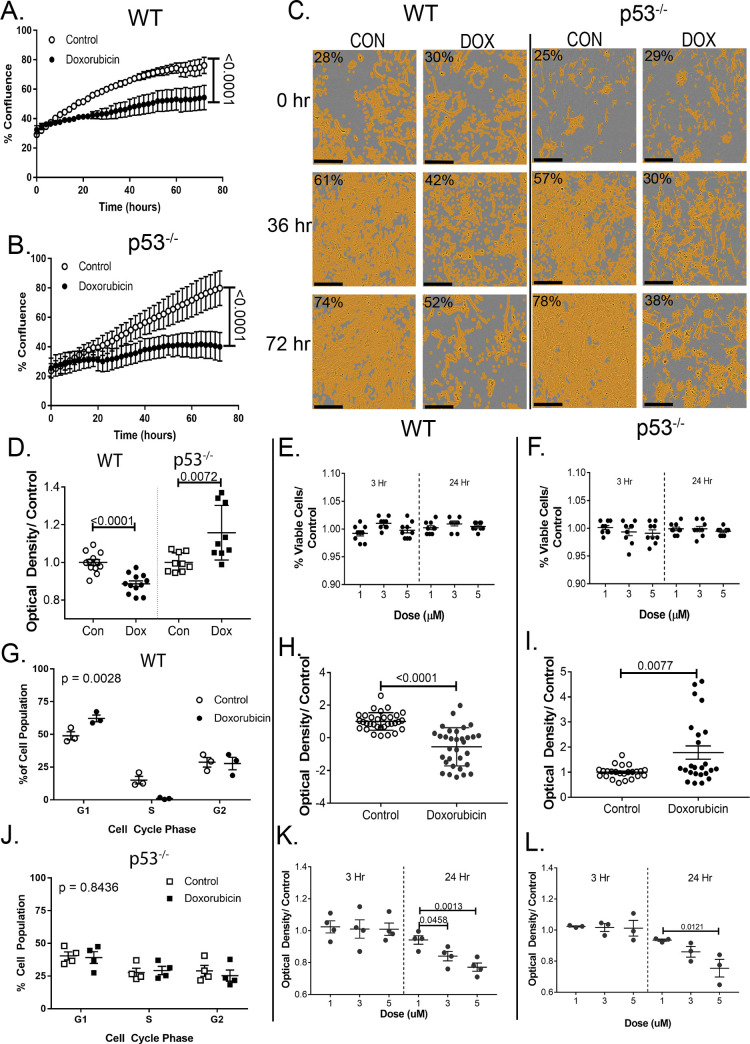
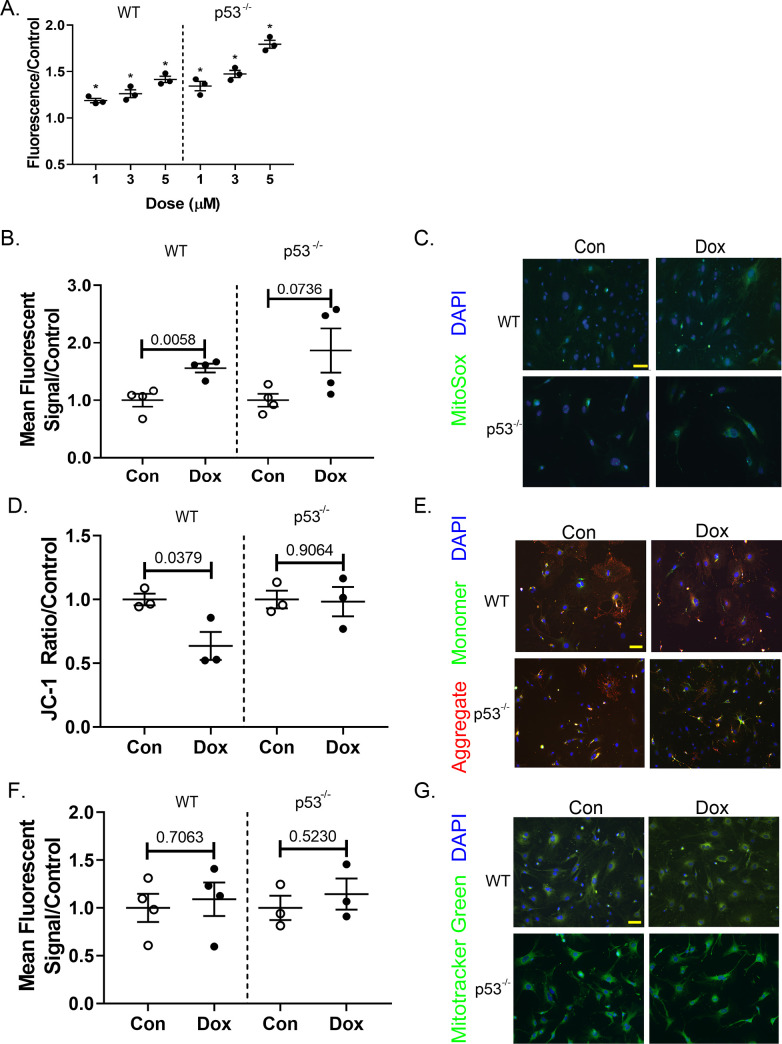
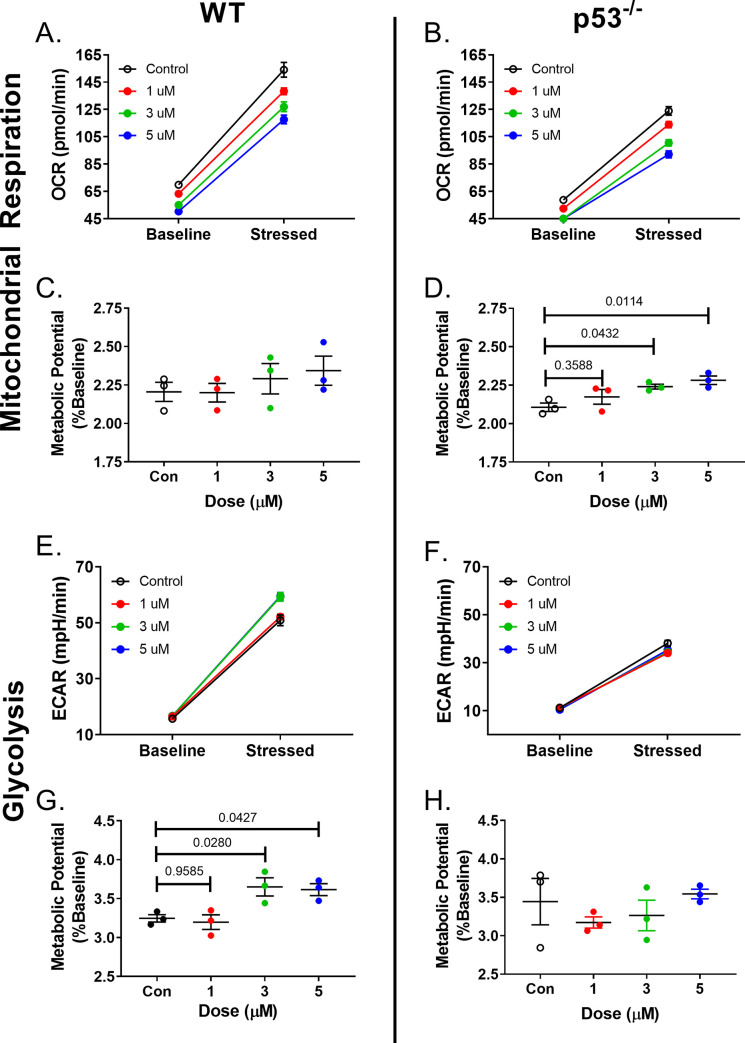
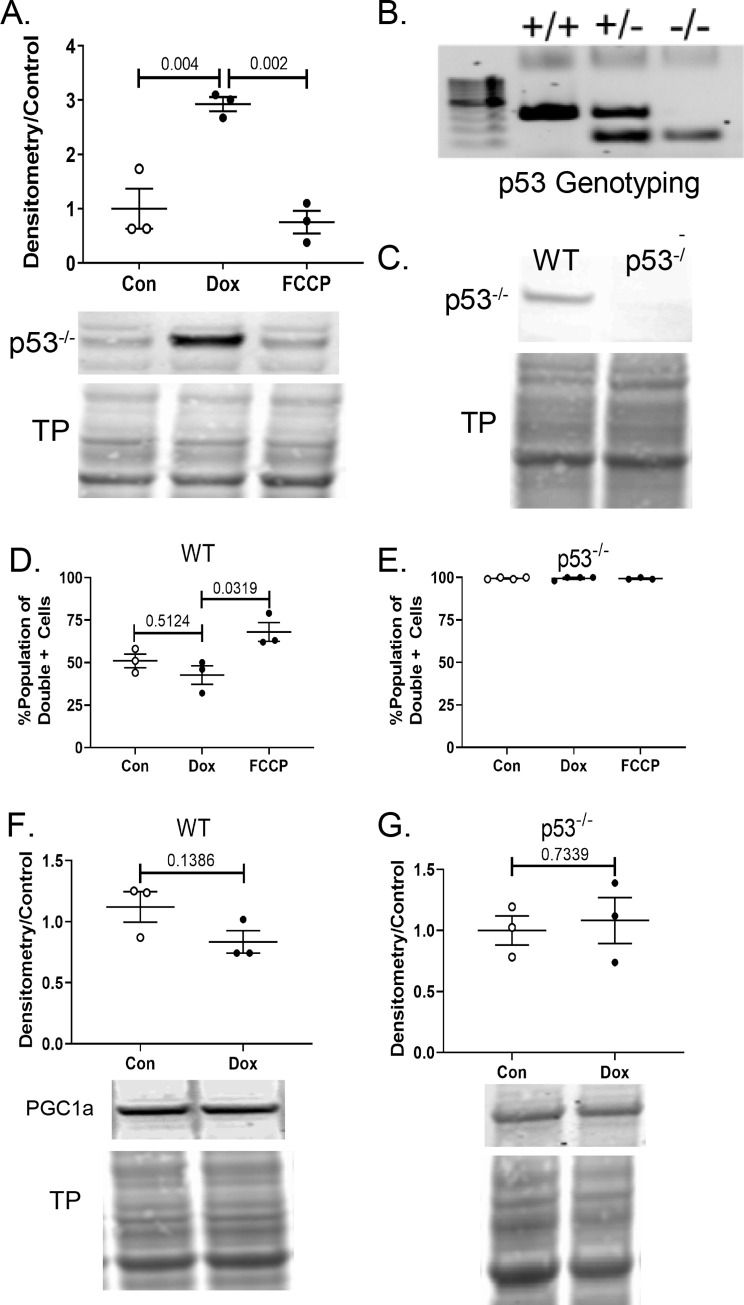
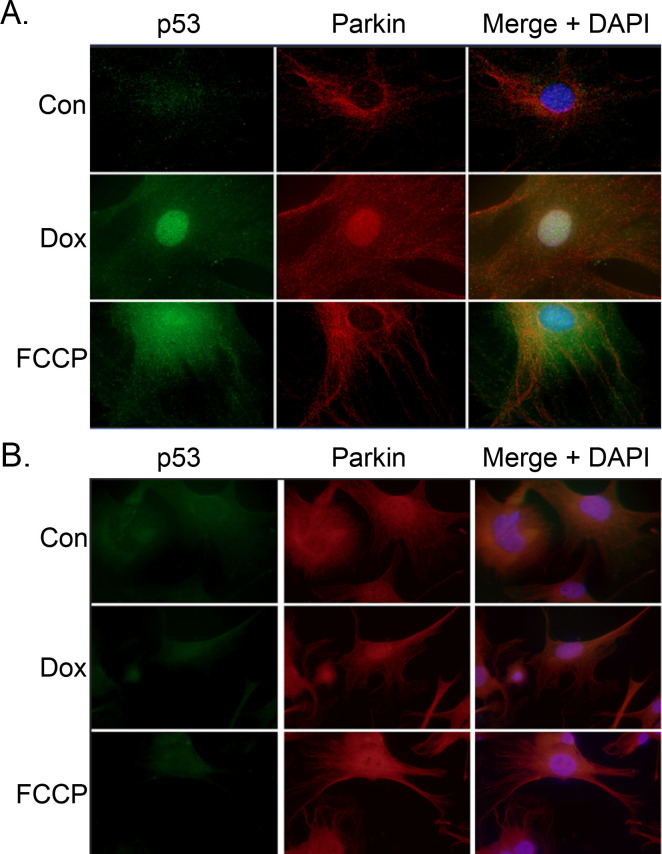
Anthracyclines are the critical component in a majority of pediatric chemotherapy regimens due to their broad anticancer efficacy. Unfortunately, the vast majority of long-term childhood cancer survivors will develop a chronic health condition caused by their successful treatments and severe cardiac disease is a common life-threatening outcome that is unequivocally linked to previous anthracycline exposure. The intricacies of how anthracyclines such as doxorubicin, damage the heart and initiate a disease process that progresses over multiple decades is not fully understood. One area left largely unstudied is the role of the cardiac fibroblast, a key cell type in cardiac maturation and injury response. In this study, we demonstrate the effect of doxorubicin on cardiac fibroblast function in the presence and absence of the critical DNA damage response protein p53. In wildtype cardiac fibroblasts, doxorubicin-induced damage correlated with decreased proliferation and migration, cell cycle arrest, and a dilated cardiomyopathy gene expression profile. Interestingly, these doxorubicin-induced changes were completely or partially restored in p53-/- cardiac fibroblasts. Moreover, in wildtype cardiac fibroblasts, doxorubicin produced DNA damage and mitochondrial dysfunction, both of which are well-characterized cell stress responses induced by cytotoxic chemotherapy and varied forms of heart injury. A 3-fold increase in p53 (p = 0.004) prevented the completion of mitophagy (p = 0.032) through sequestration of Parkin. Interactions between p53 and Parkin increased in doxorubicin-treated cardiac fibroblasts (p = 0.0003). Finally, Parkin was unable to localize to the mitochondria in wildtype cardiac fibroblasts, but mitochondrial localization was restored in p53-/- cardiac fibroblasts. These findings strongly suggest that cardiac fibroblasts are an important myocardial cell type that merits further study in the context of doxorubicin treatment. A more robust knowledge of the role cardiac fibroblasts play in the development of doxorubicin-induced cardiotoxicity will lead to novel clinical strategies that will improve the quality of life of cancer survivors.
Conflict of interest statement
NO authors have competing interest.
Figures


References
-
- Howlader N NA, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, et al. SEER Cancer Statistics Review, 1975–2012. Bethesda, MD: National Cancer Institute, 2015.
-
- Armstrong GT, Kawashima T, Leisenring W, Stratton K, Stovall M, Hudson MM, et al. Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32(12):1218–27. Epub 2014/03/19. 10.1200/jco.2013.51.1055 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
