

Efficient formulation of polarizable Gaussian multipole electrostatics for biomolecular simulations
- PMID: 32962395
- PMCID: PMC7502018
- DOI: 10.1063/5.0019560
Efficient formulation of polarizable Gaussian multipole electrostatics for biomolecular simulations
Abstract
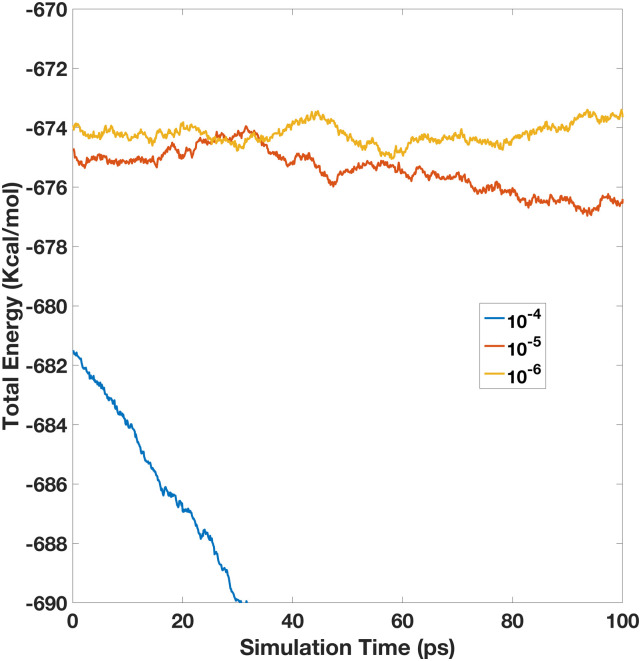
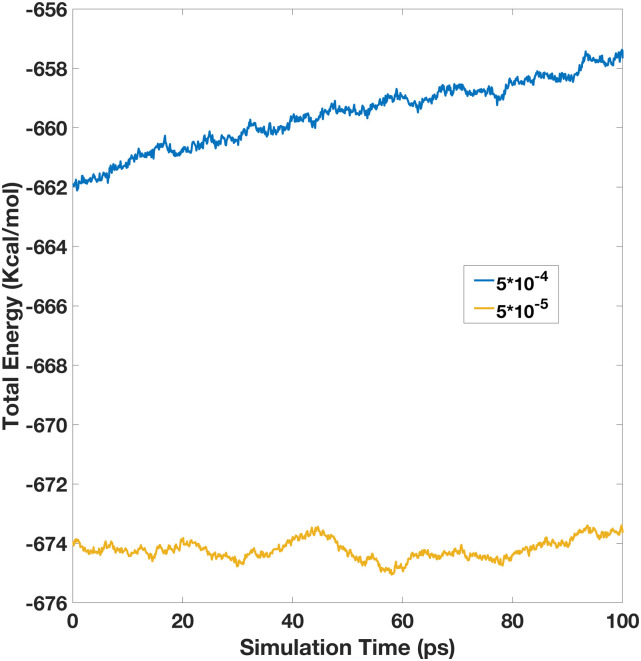
Molecular dynamics simulations of biomolecules have been widely adopted in biomedical studies. As classical point-charge models continue to be used in routine biomolecular applications, there have been growing demands on developing polarizable force fields for handling more complicated biomolecular processes. Here, we focus on a recently proposed polarizable Gaussian Multipole (pGM) model for biomolecular simulations. A key benefit of pGM is its screening of all short-range electrostatic interactions in a physically consistent manner, which is critical for stable charge-fitting and is needed to reproduce molecular anisotropy. Another advantage of pGM is that each atom's multipoles are represented by a single Gaussian function or its derivatives, allowing for more efficient electrostatics than other Gaussian-based models. In this study, we present an efficient formulation for the pGM model defined with respect to a local frame formed with a set of covalent basis vectors. The covalent basis vectors are chosen to be along each atom's covalent bonding directions. The new local frame can better accommodate the fact that permanent dipoles are primarily aligned along covalent bonds due to the differences in electronegativity of bonded atoms. It also allows molecular flexibility during molecular simulations and facilitates an efficient formulation of analytical electrostatic forces without explicit torque computation. Subsequent numerical tests show that analytical atomic forces agree excellently with numerical finite-difference forces for the tested system. Finally, the new pGM electrostatics algorithm is interfaced with the particle mesh Ewald (PME) implementation in Amber for molecular simulations under the periodic boundary conditions. To validate the overall pGM/PME electrostatics, we conducted an NVE simulation for a small water box of 512 water molecules. Our results show that to achieve energy conservation in the polarizable model, it is important to ensure enough accuracy on both PME and induction iteration. It is hoped that the reformulated pGM model will facilitate the development of future force fields based on the pGM electrostatics for applications in biomolecular systems and processes where polarization plays crucial roles.
Figures
References
-
- Burkert U. and Allinger N., Molecular Mechanics, ACS Monograph Series (American Chemical Society, Washington, DC, 1982).
-
- Jorgensen W. L. et al., J. Chem. Phys. 79, 926 (1983).10.1063/1.445869 - DOI
-
- Berendsen H. J. et al., Intermolecular Forces (Springer, 1981), p. 331.
-
- Mahoney M. W. and Jorgensen W. L., J. Chem. Phys. 112, 8910 (2000).10.1063/1.481505 - DOI
-
- Clementi E. et al., Theor. Chim. Acta 55, 257 (1980).10.1007/bf00549424 - DOI