

Identification of a SCN4A mutation in a large Chinese family with atypical normokalemic periodic paralysis using whole-exome sequencing
- PMID: 32962503
- PMCID: PMC7517994
- DOI: 10.1177/0300060520953643
Identification of a SCN4A mutation in a large Chinese family with atypical normokalemic periodic paralysis using whole-exome sequencing
Abstract
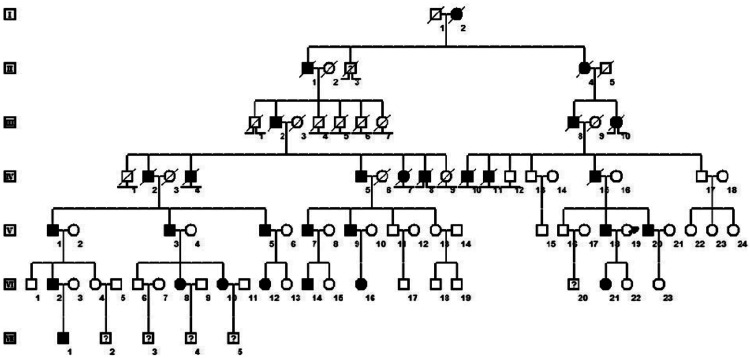
Objectives: Normokalemic periodic paralysis (NormoKPP) of skeletal muscle is an autosomal dominant disorder caused by mutations in the gene encoding voltage-gated sodium channel protein type 4 subunit alpha (SCN4A), which leads to ion channel dysfunction. Little is known about the relationship between genotype and the clinical symptoms of NormoKPP. The present study aimed to evaluate the genetic variation in a large Chinese family with NormoKPP. The patients in this pedigree did not respond to saline treatment, but calcium gluconate treatment was effective.
Methods: We performed a series of clinical examinations and genetic analyses, using whole-exome and Sanger sequencing, to examine the mutation status of SCN4A in a Chinese family segregating for NormoKPP.
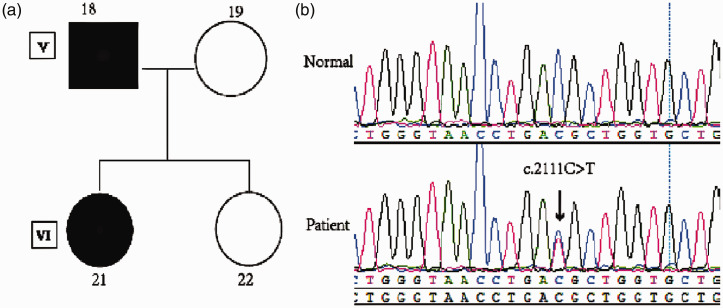
Results: Whole-exome sequencing revealed a c.2111C>T substitution in SCN4A in most of the affected family members. This mutation results in the amino acid substitution p.T704M.
Conclusions: These results support a causative role of this mutation in SCN4A in NormoKPP, and provide information about the relationship between genotype and atypical clinical symptoms.
Keywords: Normokalemic periodic paralysis; SCN4A; Sanger sequencing; ion channel; mutation; whole-exome sequencing.
Figures
Similar articles
-
[Clinical and molecular genetic analysis of a family with normokalemic periodic paralysis].Zhonghua Er Ke Za Zhi. 2013 Jan;51(1):47-51. Zhonghua Er Ke Za Zhi. 2013. PMID: 23527931 Chinese.
-
Successful treatment of normokalemic periodic paralysis with hydrochlorothiazide.Brain Dev. 2018 Oct;40(9):833-836. doi: 10.1016/j.braindev.2018.05.011. Epub 2018 Jun 12. Brain Dev. 2018. PMID: 29907477
-
Normokalemic periodic paralysis is not a distinct disease.Muscle Nerve. 2012 Dec;46(6):914-6. doi: 10.1002/mus.23441. Epub 2012 Aug 24. Muscle Nerve. 2012. PMID: 22926674
-
Overlap of periodic paralysis and paramyotonia congenita caused by SCN4A gene mutations two family reports and literature review.Channels (Austin). 2019 Dec;13(1):110-119. doi: 10.1080/19336950.2019.1600967. Channels (Austin). 2019. PMID: 30931713 Free PMC article. Review.
-
Mutations of SCN4A gene cause different diseases: 2 case reports and literature review.Channels (Austin). 2015;9(2):82-7. doi: 10.1080/19336950.2015.1012945. Channels (Austin). 2015. PMID: 25839108 Free PMC article. Review.
Cited by
-
Hypokalemic Periodic Paralysis Type 2 Due to SCN4A Val1105Met Mutation: A Case Study.Cureus. 2024 Jan 10;16(1):e52063. doi: 10.7759/cureus.52063. eCollection 2024 Jan. Cureus. 2024. PMID: 38344586 Free PMC article.
References
-
- Lehmann-Horn F, Küther G, Ricker K, et al. Adynamia episodica hereditaria with myotonia: a noninactivating sodium current and the effect of extracellular pH. Muscle Nerve 1987; 10: 363–374. - PubMed
-
- Bulman DE, Scoggan KA, Van Oene MD, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 1999; 53: 1932–1936. - PubMed
-
- Fontaine B, Vale-Santos J, Jurkat-Rott K, et al. Mapping of the hypokalaemic periodic paralysis (HypoPP) locus to chromosome 1q31-32 in three European families. Nat Genet 1994; 6: 267–272. - PubMed
-
- Vicart S, Sternberg D, Fournier E, et al. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology 2004; 63: 2120–2127. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
