

Assessment of Multilocus Sequence Analysis (MLSA) for Identification of Candidatus Liberibacter Solanacearum from Different Host Plants in Spain
- PMID: 32967215
- PMCID: PMC7565762
- DOI: 10.3390/microorganisms8091446
Assessment of Multilocus Sequence Analysis (MLSA) for Identification of Candidatus Liberibacter Solanacearum from Different Host Plants in Spain
Abstract
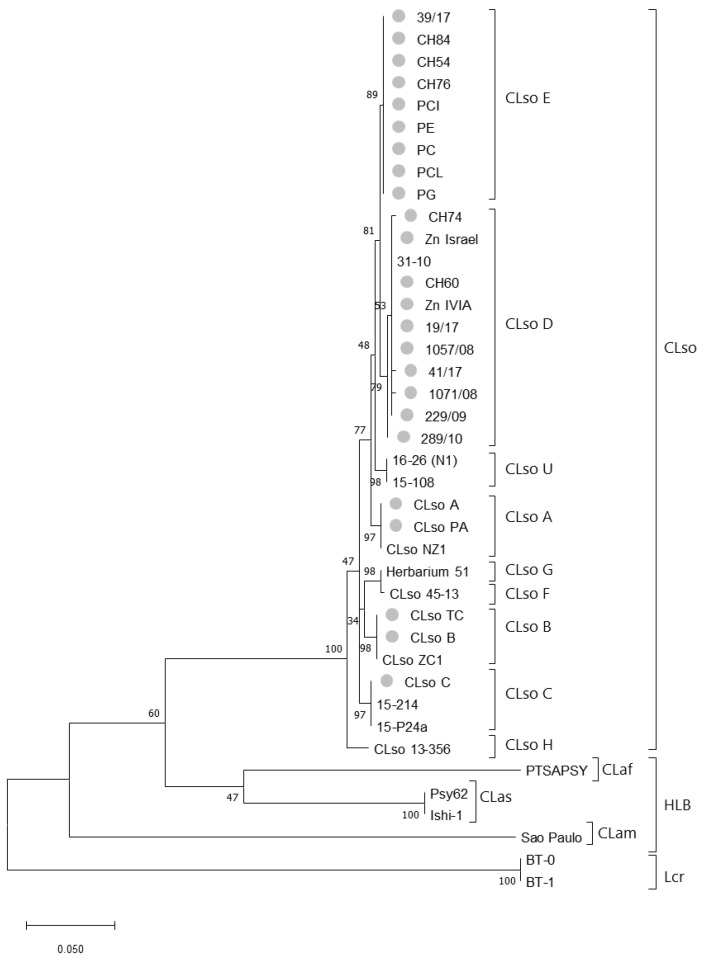
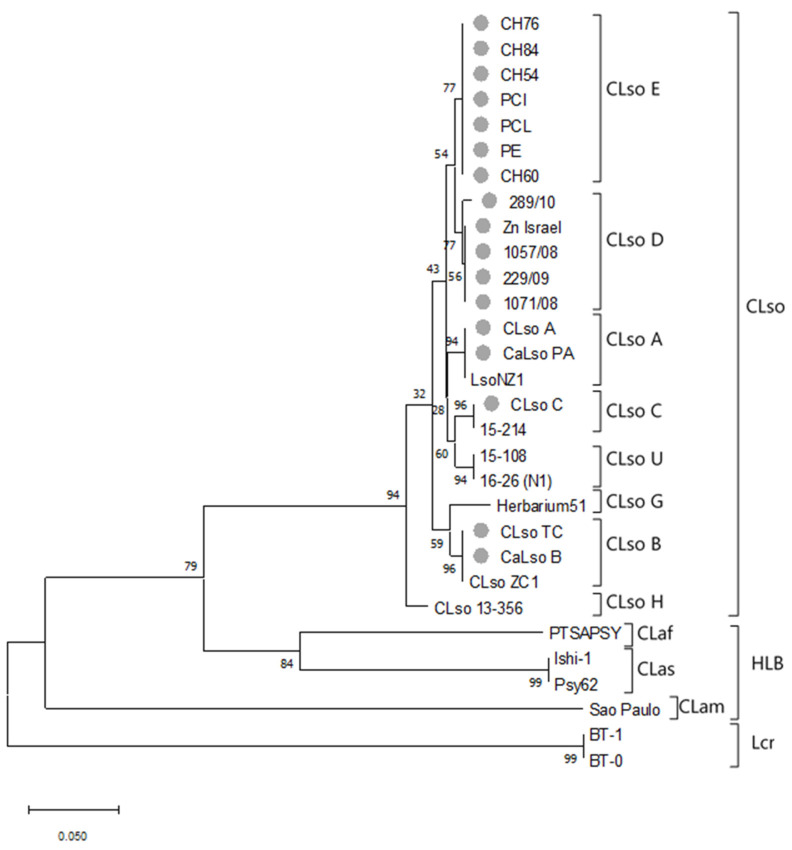
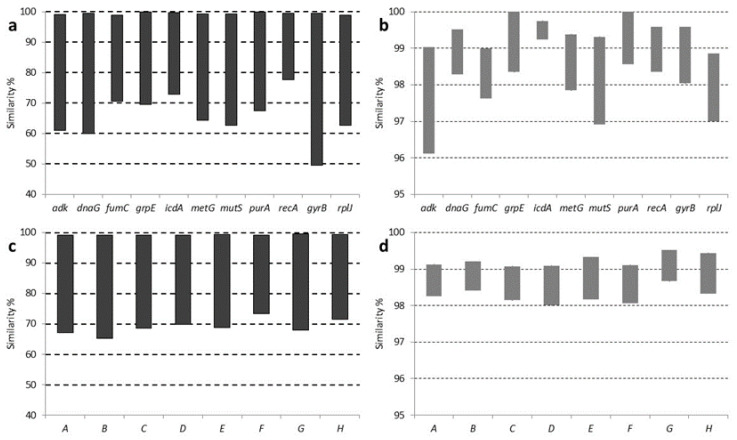
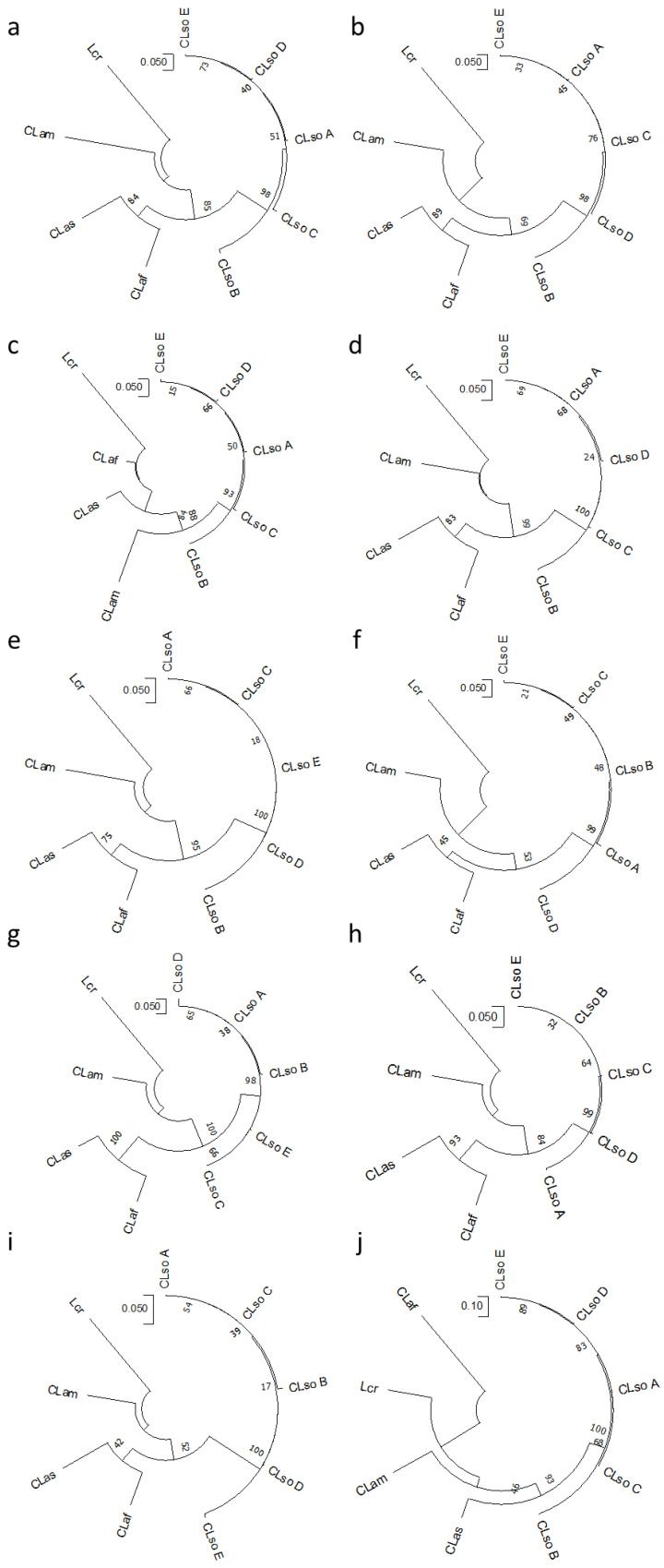
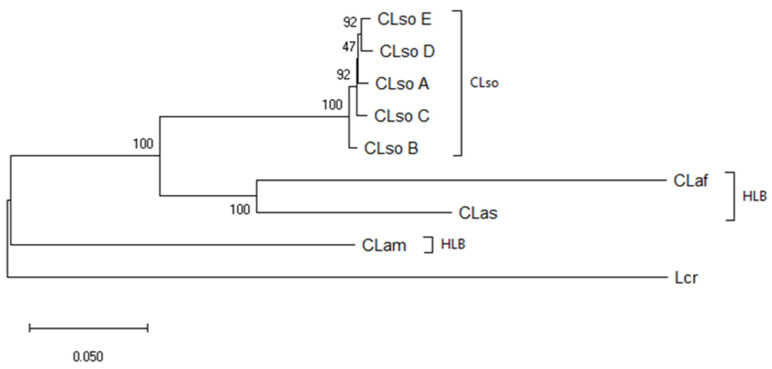
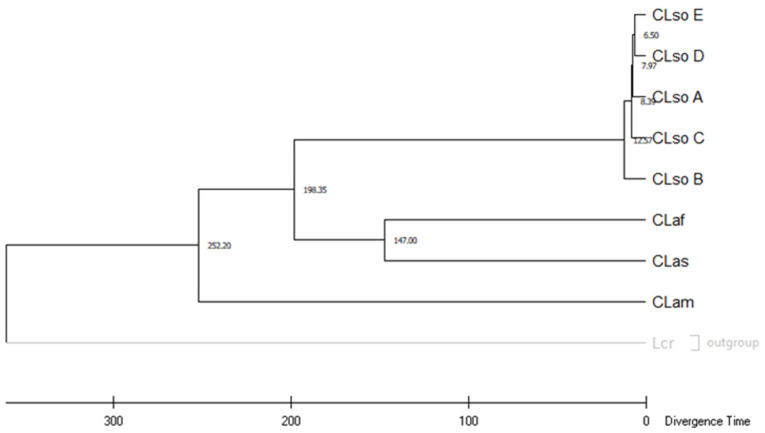
Liberibacter is a bacterial group causing different diseases and disorders in plants. Among liberibacters, Candidatus Liberibacter solanaceraum (CLso) produces disorders in several species mainly within Apiaceae and Solanaceae families. CLso isolates are usually grouped in defined haplotypes according to single nucleotide polymorphisms in genes associated with ribosomal elements. In order to characterize more precisely isolates of CLso identified in potato in Spain, a Multilocus Sequence Analysis (MLSA) was applied. This methodology was validated by a complete analysis of ten housekeeping genes that showed an absence of positive selection and a nearly neutral mechanism for their evolution. Most of the analysis performed with single housekeeping genes, as well as MLSA, grouped together isolates of CLso detected in potato crops in Spain within the haplotype E, undistinguishable from those infecting carrots, parsnips or celery. Moreover, the information from these housekeeping genes was used to estimate the evolutionary divergence among the different CLso by using the concatenated sequences of the genes assayed. Data obtained on the divergence among CLso haplotypes support the hypothesis of evolutionary events connected with different hosts, in different geographic areas, and possibly associated with different vectors. Our results demonstrate the absence in Spain of CLso isolates molecularly classified as haplotypes A and B, traditionally considered causal agents of zebra chip in potato, as well as the uncertain possibility of the present haplotype to produce major disease outbreaks in potato that may depend on many factors that should be further evaluated in future works.
Keywords: HLB; Liberibacter; MLSA; carrot; celery; citrus; parsnip; potato; zebra chip.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Conventional and qPCR reveals the presence of 'Candidatus Liberibacter solanacearum' haplotypes A, and B in Physalis philadelphica plant, seed, and Βactericera cockerelli psyllids, with the assignment of a new haplotype H in Convolvulaceae.Antonie Van Leeuwenhoek. 2020 Apr;113(4):533-551. doi: 10.1007/s10482-019-01362-9. Epub 2019 Nov 27. Antonie Van Leeuwenhoek. 2020. PMID: 31776768
-
Genetic Variation of 'Candidatus Liberibacter solanacearum' Haplotype C and Identification of a Novel Haplotype from Trioza urticae and Stinging Nettle.Phytopathology. 2018 Aug;108(8):925-934. doi: 10.1094/PHYTO-12-17-0410-R. Epub 2018 Jun 20. Phytopathology. 2018. PMID: 29600888
-
The functional decline of tomato plants infected by Candidatus Liberbacter solanacearum: an RNA-seq transcriptomic analysis.Front Plant Sci. 2024 Feb 1;15:1325254. doi: 10.3389/fpls.2024.1325254. eCollection 2024. Front Plant Sci. 2024. PMID: 38362455 Free PMC article.
-
The Candidatus Liberibacter-Host Interface: Insights into Pathogenesis Mechanisms and Disease Control.Annu Rev Phytopathol. 2017 Aug 4;55:451-482. doi: 10.1146/annurev-phyto-080516-035513. Epub 2017 Jun 21. Annu Rev Phytopathol. 2017. PMID: 28637377 Review.
-
Progress and Obstacles in Culturing 'Candidatus Liberibacter asiaticus', the Bacterium Associated with Huanglongbing.Phytopathology. 2019 Jul;109(7):1092-1101. doi: 10.1094/PHYTO-02-19-0051-RVW. Epub 2019 Jun 3. Phytopathology. 2019. PMID: 30998129 Review.
Cited by
-
Survey of Candidatus Liberibacter Solanacearum and Its Associated Vectors in Potato Crop in Spain.Insects. 2022 Oct 21;13(10):964. doi: 10.3390/insects13100964. Insects. 2022. PMID: 36292913 Free PMC article.
-
Reference-Free Plant Disease Detection Using Machine Learning and Long-Read Metagenomic Sequencing.Appl Environ Microbiol. 2023 Jun 28;89(6):e0026023. doi: 10.1128/aem.00260-23. Epub 2023 May 15. Appl Environ Microbiol. 2023. PMID: 37184398 Free PMC article.
-
Genetic diversity of microsymbionts nodulating Trifolium pratense in subpolar and temperate climate regions.Sci Rep. 2022 Jul 15;12(1):12144. doi: 10.1038/s41598-022-16410-0. Sci Rep. 2022. PMID: 35840628 Free PMC article.
-
Assessment of Psyllid Handling and DNA Extraction Methods in the Detection of 'Candidatus Liberibacter Solanacearum' by qPCR.Microorganisms. 2022 May 26;10(6):1104. doi: 10.3390/microorganisms10061104. Microorganisms. 2022. PMID: 35744622 Free PMC article.
References
-
- Haapalainen M. Biology and epidemics of Candidatus Liberibacter species, psyllid-transmitted plant-pathogenic bacteria. Ann. Appl. Biol. 2014;165:172–198. doi: 10.1111/aab.12149. - DOI
-
- Raddadi N., Gonella E., Camerota C., Pizzinat A., Tedeschi R., Crotti E., Mandrioli M., Attilio Bianco P., Daffonchio D., Alma A. ‘Candidatus Liberibacter europaeus’ sp. nov. that is associated with and transmitted by the psyllid Cacopsylla pyri apparently behaves as an endophyte rather than a pathogen. Environ. Microbiol. 2011;13:414–426. doi: 10.1111/j.1462-2920.2010.02347.x. - DOI - PubMed
-
- Alfaro-Fernández A., Hernández-Llopis D., Font M.I. Haplotypes of ‘Candidatus Liberibacter solanacearum’ identified in Umbeliferous crops in Spain. Eur. J. Plant Pathol. 2017;149:127–131. doi: 10.1007/s10658-017-1172-2. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
