

Ensemble-based enzyme design can recapitulate the effects of laboratory directed evolution in silico
- PMID: 32968058
- PMCID: PMC7511930
- DOI: 10.1038/s41467-020-18619-x
Ensemble-based enzyme design can recapitulate the effects of laboratory directed evolution in silico
Abstract
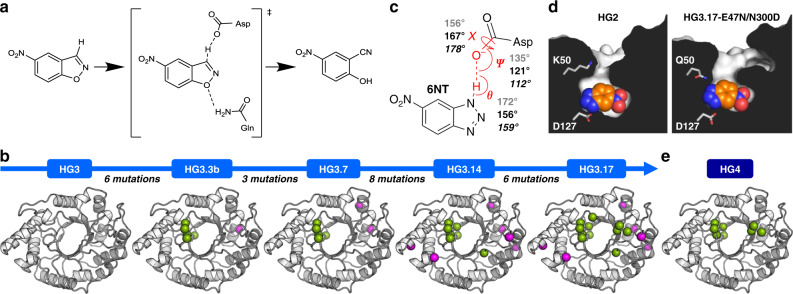
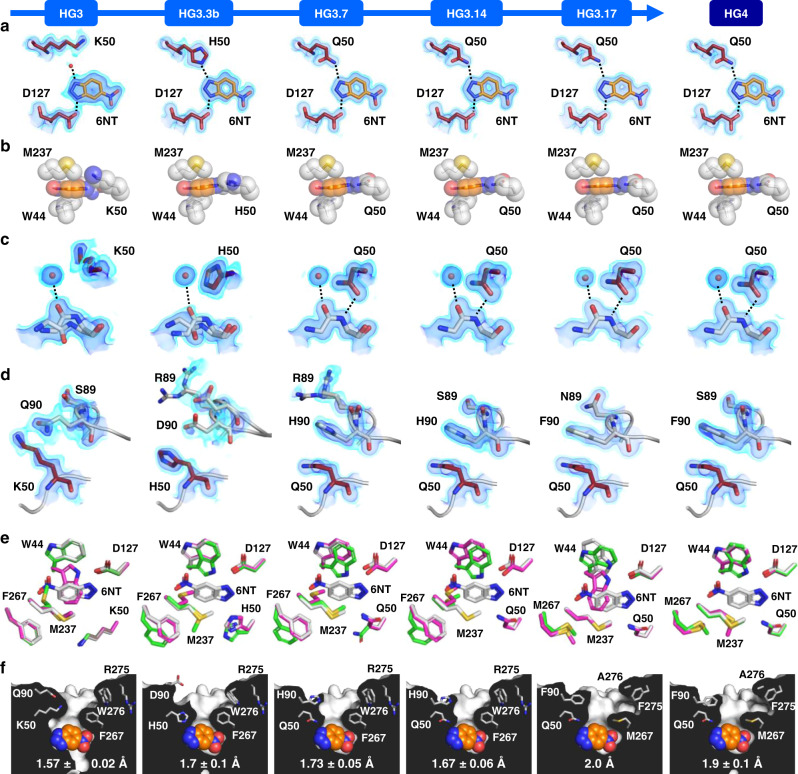
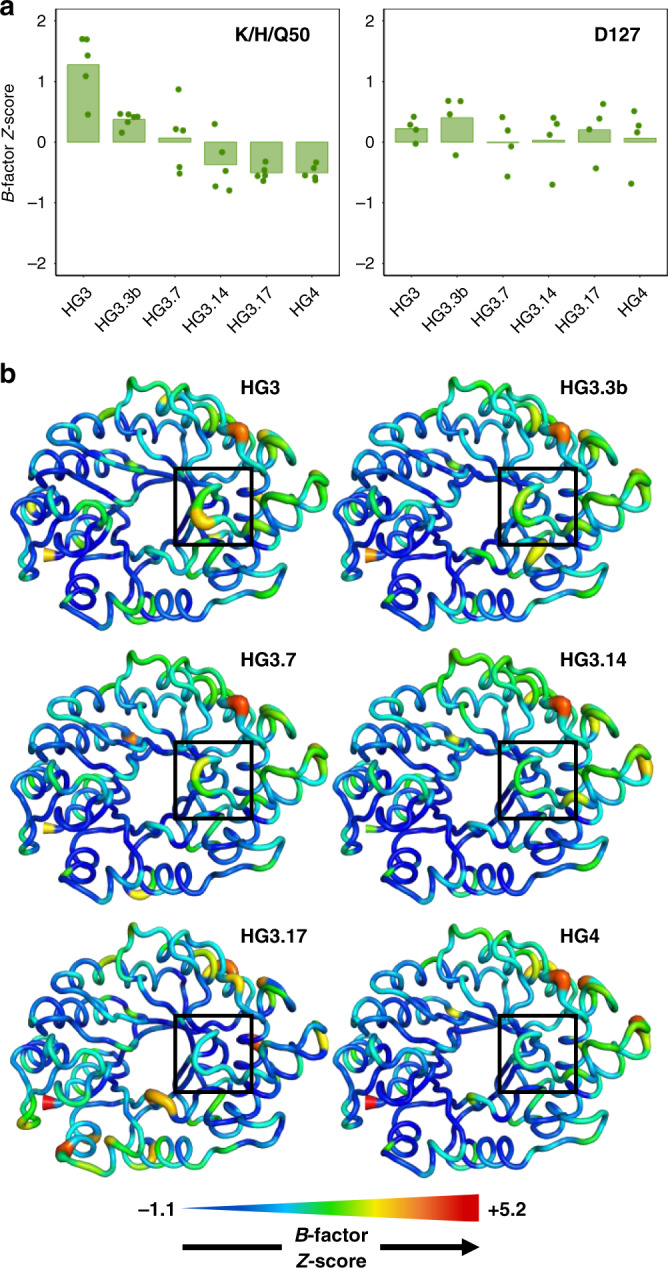
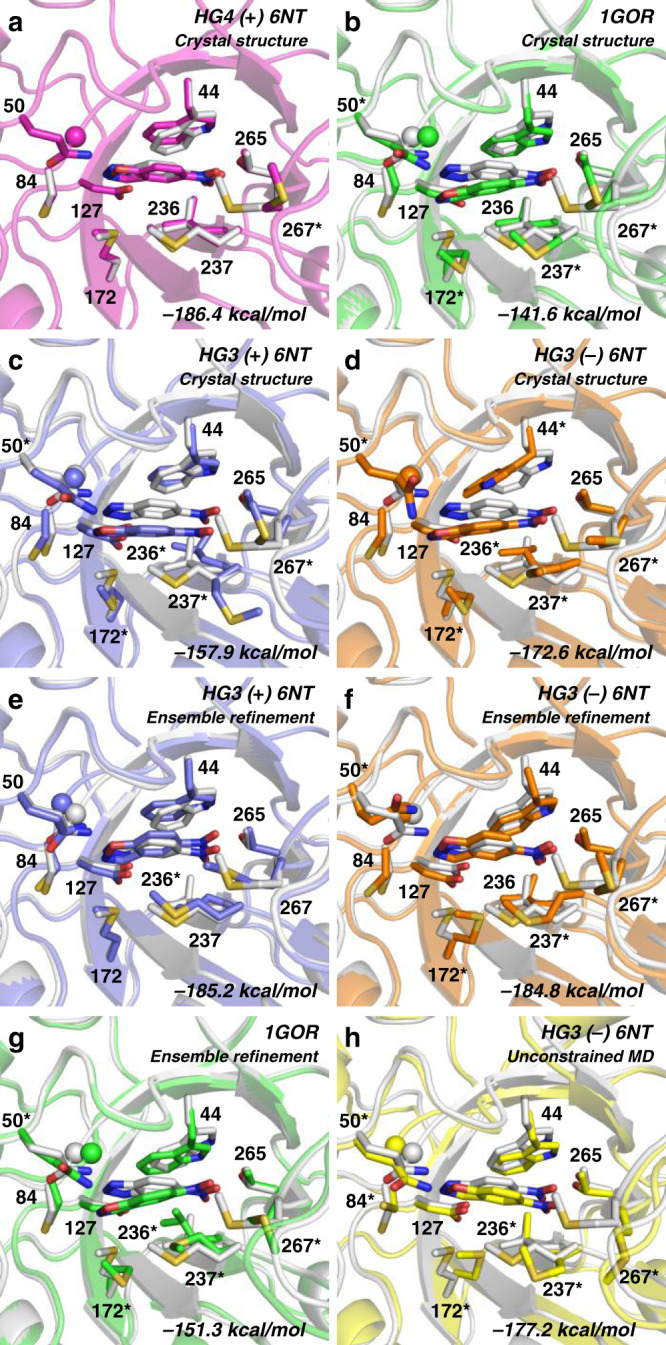
The creation of artificial enzymes is a key objective of computational protein design. Although de novo enzymes have been successfully designed, these exhibit low catalytic efficiencies, requiring directed evolution to improve activity. Here, we use room-temperature X-ray crystallography to study changes in the conformational ensemble during evolution of the designed Kemp eliminase HG3 (kcat/KM 146 M-1s-1). We observe that catalytic residues are increasingly rigidified, the active site becomes better pre-organized, and its entrance is widened. Based on these observations, we engineer HG4, an efficient biocatalyst (kcat/KM 103,000 M-1s-1) containing key first and second-shell mutations found during evolution. HG4 structures reveal that its active site is pre-organized and rigidified for efficient catalysis. Our results show how directed evolution circumvents challenges inherent to enzyme design by shifting conformational ensembles to favor catalytically-productive sub-states, and suggest improvements to the design methodology that incorporate ensemble modeling of crystallographic data.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
