

An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I
- PMID: 32971903
- PMCID: PMC7564762
- DOI: 10.3390/genes11091109
An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I
Abstract
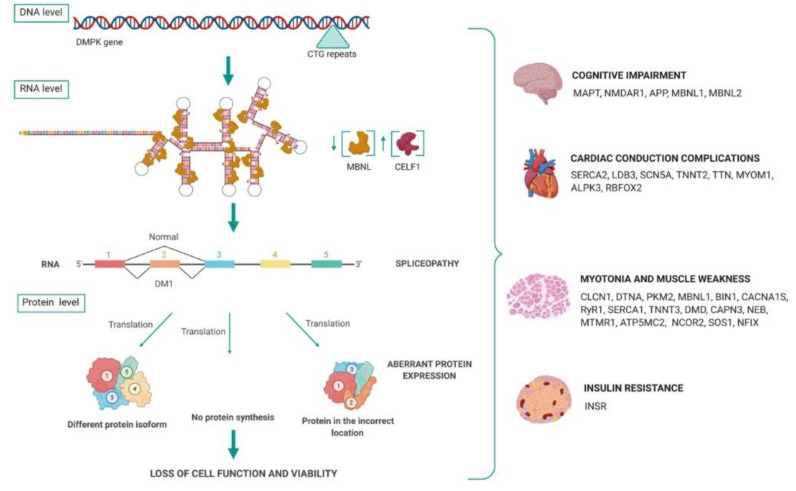
Myotonic dystrophy type I (DM1) is the most common form of adult muscular dystrophy, caused by expansion of a CTG triplet repeat in the 3' untranslated region (3'UTR) of the myotonic dystrophy protein kinase (DMPK) gene. The pathological CTG repeats result in protein trapping by expanded transcripts, a decreased DMPK translation and the disruption of the chromatin structure, affecting neighboring genes expression. The muscleblind-like (MBNL) and CUG-BP and ETR-3-like factors (CELF) are two families of tissue-specific regulators of developmentally programmed alternative splicing that act as antagonist regulators of several pre-mRNA targets, including troponin 2 (TNNT2), insulin receptor (INSR), chloride channel 1 (CLCN1) and MBNL2. Sequestration of MBNL proteins and up-regulation of CELF1 are key to DM1 pathology, inducing a spliceopathy that leads to a developmental remodelling of the transcriptome due to an adult-to-foetal splicing switch, which results in the loss of cell function and viability. Moreover, recent studies indicate that additional pathogenic mechanisms may also contribute to disease pathology, including a misregulation of cellular mRNA translation, localization and stability. This review focuses on the cause and effects of MBNL and CELF1 deregulation in DM1, describing the molecular mechanisms underlying alternative splicing misregulation for a deeper understanding of DM1 complexity. To contribute to this analysis, we have prepared a comprehensive list of transcript alterations involved in DM1 pathogenesis, as well as other deregulated mRNA processing pathways implications.
Keywords: CELF1; DMPK; MBNL; myotonic dystrophy; spliceopathy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The hallmarks of myotonic dystrophy type 1 muscle dysfunction.Biol Rev Camb Philos Soc. 2021 Apr;96(2):716-730. doi: 10.1111/brv.12674. Epub 2020 Dec 2. Biol Rev Camb Philos Soc. 2021. PMID: 33269537 Review.
-
Sense and Antisense DMPK RNA Foci Accumulate in DM1 Tissues during Development.PLoS One. 2015 Sep 4;10(9):e0137620. doi: 10.1371/journal.pone.0137620. eCollection 2015. PLoS One. 2015. PMID: 26339785 Free PMC article.
-
Choroid plexus mis-splicing and altered cerebrospinal fluid composition in myotonic dystrophy type 1.Brain. 2023 Oct 3;146(10):4217-4232. doi: 10.1093/brain/awad148. Brain. 2023. PMID: 37143315 Free PMC article.
-
(CTG)n repeat-mediated dysregulation of MBNL1 and MBNL2 expression during myogenesis in DM1 occurs already at the myoblast stage.PLoS One. 2019 May 22;14(5):e0217317. doi: 10.1371/journal.pone.0217317. eCollection 2019. PLoS One. 2019. PMID: 31116797 Free PMC article.
-
Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics.Dev Dyn. 2015 Mar;244(3):377-90. doi: 10.1002/dvdy.24240. Epub 2015 Jan 13. Dev Dyn. 2015. PMID: 25504326 Review.
Cited by
-
Peptide-conjugated antimiRs improve myotonic dystrophy type 1 phenotypes by promoting endogenous MBNL1 expression.Mol Ther Nucleic Acids. 2023 Sep 5;34:102024. doi: 10.1016/j.omtn.2023.09.001. eCollection 2023 Dec 12. Mol Ther Nucleic Acids. 2023. PMID: 37744174 Free PMC article.
-
Cellular Senescence and Aging in Myotonic Dystrophy.Int J Mol Sci. 2022 Feb 20;23(4):2339. doi: 10.3390/ijms23042339. Int J Mol Sci. 2022. PMID: 35216455 Free PMC article. Review.
-
Modulatory role of RNA helicases in MBNL-dependent alternative splicing regulation.Cell Mol Life Sci. 2023 Oct 26;80(11):335. doi: 10.1007/s00018-023-04927-0. Cell Mol Life Sci. 2023. PMID: 37882878 Free PMC article.
-
Systematic Identification and Functional Validation of New snoRNAs in Human Muscle Progenitors.Noncoding RNA. 2021 Sep 13;7(3):56. doi: 10.3390/ncrna7030056. Noncoding RNA. 2021. PMID: 34564318 Free PMC article.
-
Muscle-specific gene editing improves molecular and phenotypic defects in a mouse model of myotonic dystrophy type 1.Clin Transl Med. 2025 Feb;15(2):e70227. doi: 10.1002/ctm2.70227. Clin Transl Med. 2025. PMID: 39956955 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
