

Sequence analysis of SARS-CoV-2 genome reveals features important for vaccine design
- PMID: 32973171
- PMCID: PMC7519053
- DOI: 10.1038/s41598-020-72533-2
Sequence analysis of SARS-CoV-2 genome reveals features important for vaccine design
Abstract
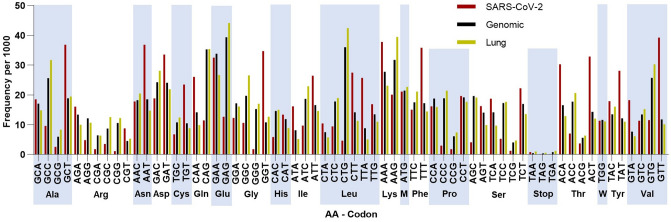
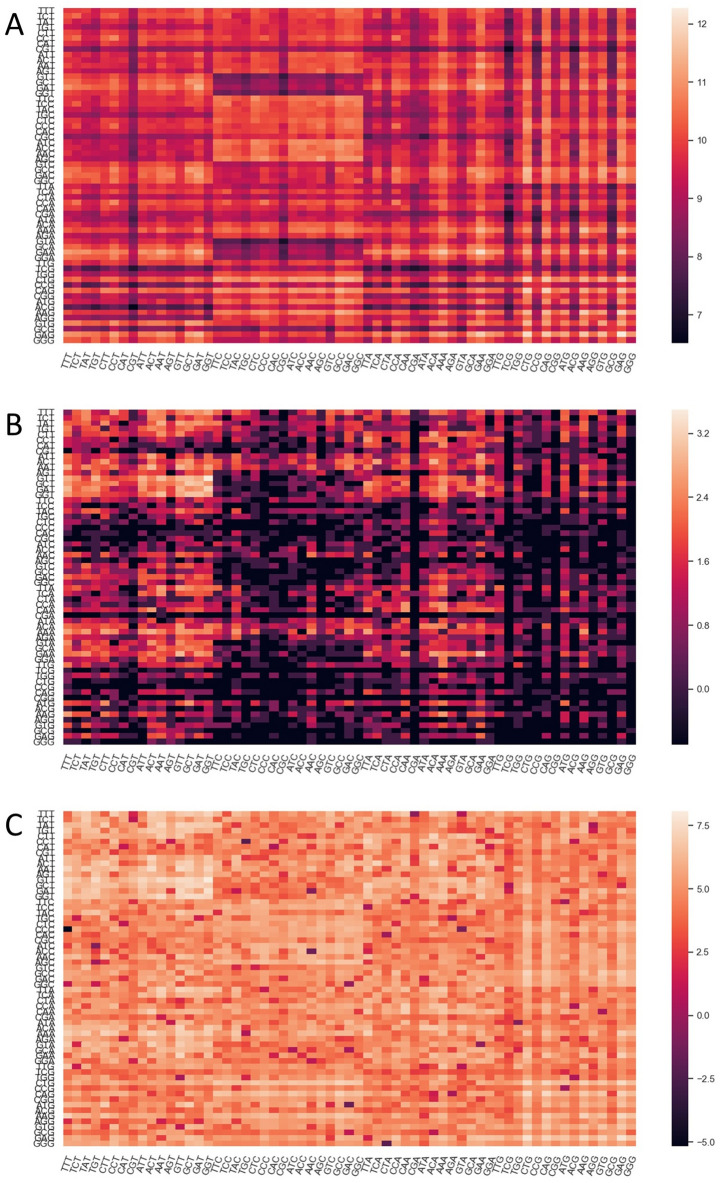
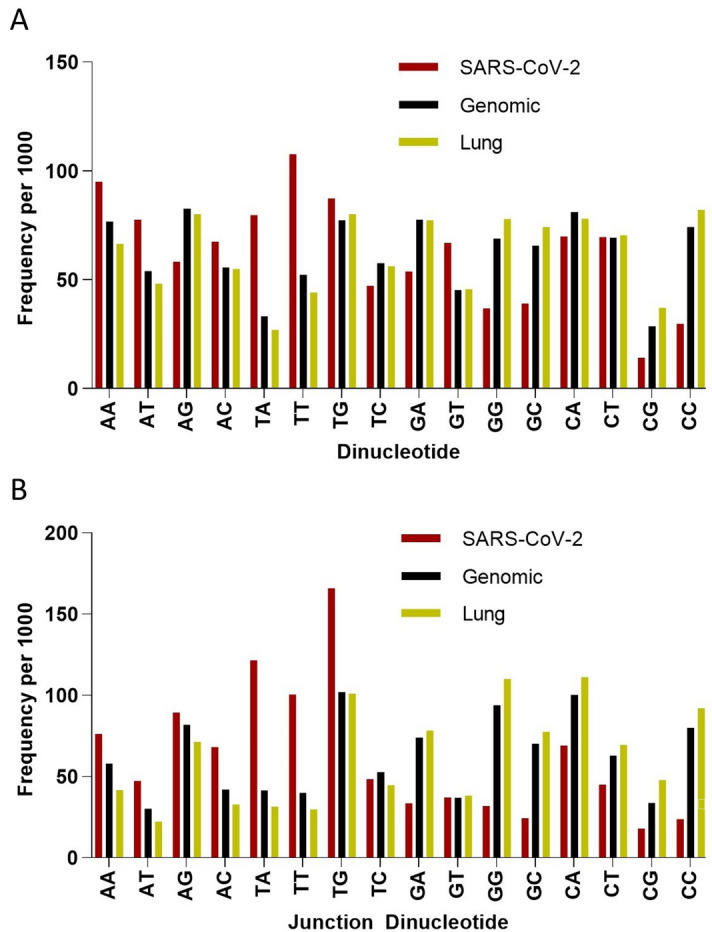
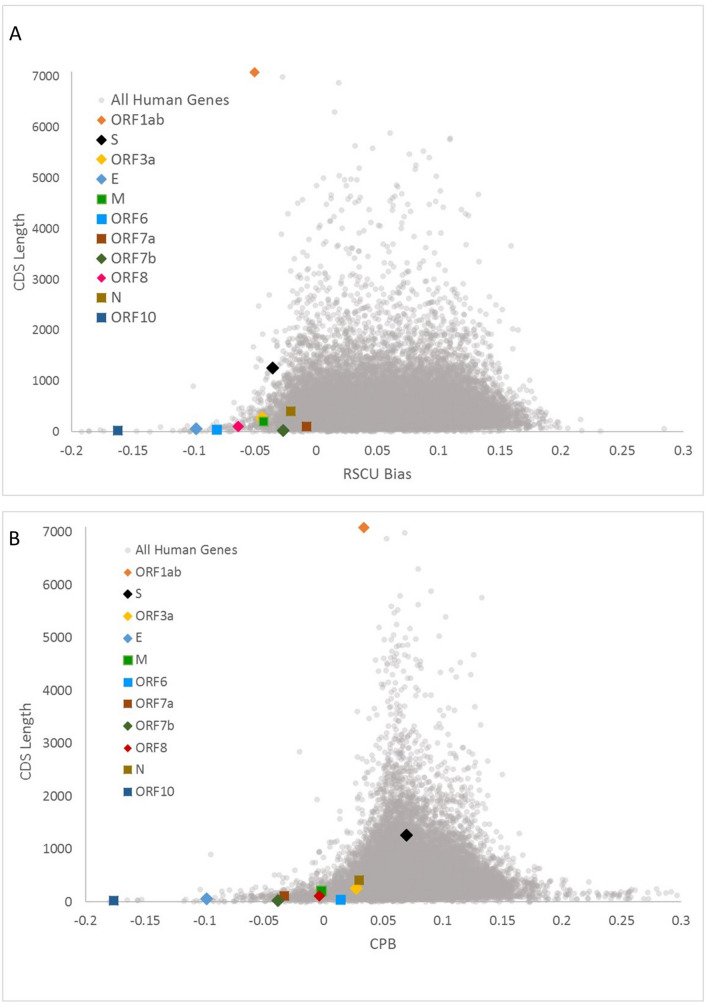
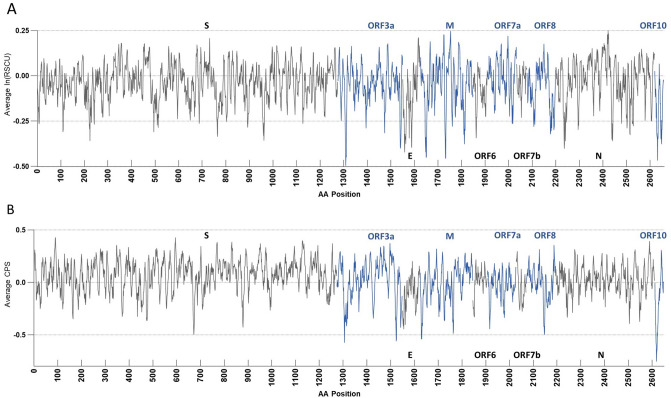
As the SARS-CoV-2 pandemic is rapidly progressing, the need for the development of an effective vaccine is critical. A promising approach for vaccine development is to generate, through codon pair deoptimization, an attenuated virus. This approach carries the advantage that it only requires limited knowledge specific to the virus in question, other than its genome sequence. Therefore, it is well suited for emerging viruses, for which we may not have extensive data. We performed comprehensive in silico analyses of several features of SARS-CoV-2 genomic sequence (e.g., codon usage, codon pair usage, dinucleotide/junction dinucleotide usage, RNA structure around the frameshift region) in comparison with other members of the coronaviridae family of viruses, the overall human genome, and the transcriptome of specific human tissues such as lung, which are primarily targeted by the virus. Our analysis identified the spike (S) and nucleocapsid (N) proteins as promising targets for deoptimization and suggests a roadmap for SARS-CoV-2 vaccine development, which can be generalizable to other viruses.
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Sequence analysis of SARS-CoV-2 genome reveals features important for vaccine design.bioRxiv [Preprint]. 2020 Mar 31:2020.03.30.016832. doi: 10.1101/2020.03.30.016832. bioRxiv. 2020. Update in: Sci Rep. 2020 Sep 24;10(1):15643. doi: 10.1038/s41598-020-72533-2. PMID: 32511300 Free PMC article. Updated. Preprint.
References
-
- Coronavirus disease 2019 (COVID-19) Situation Report–140. https://www.who.int/docs/default-source/coronaviruse/situation-reports/2... (World Health Organization, Geneva, 2020).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
