

Cholesterol Pathway Inhibition Induces TGF-β Signaling to Promote Basal Differentiation in Pancreatic Cancer
- PMID: 32976774
- PMCID: PMC7572882
- DOI: 10.1016/j.ccell.2020.08.015
Cholesterol Pathway Inhibition Induces TGF-β Signaling to Promote Basal Differentiation in Pancreatic Cancer
Abstract
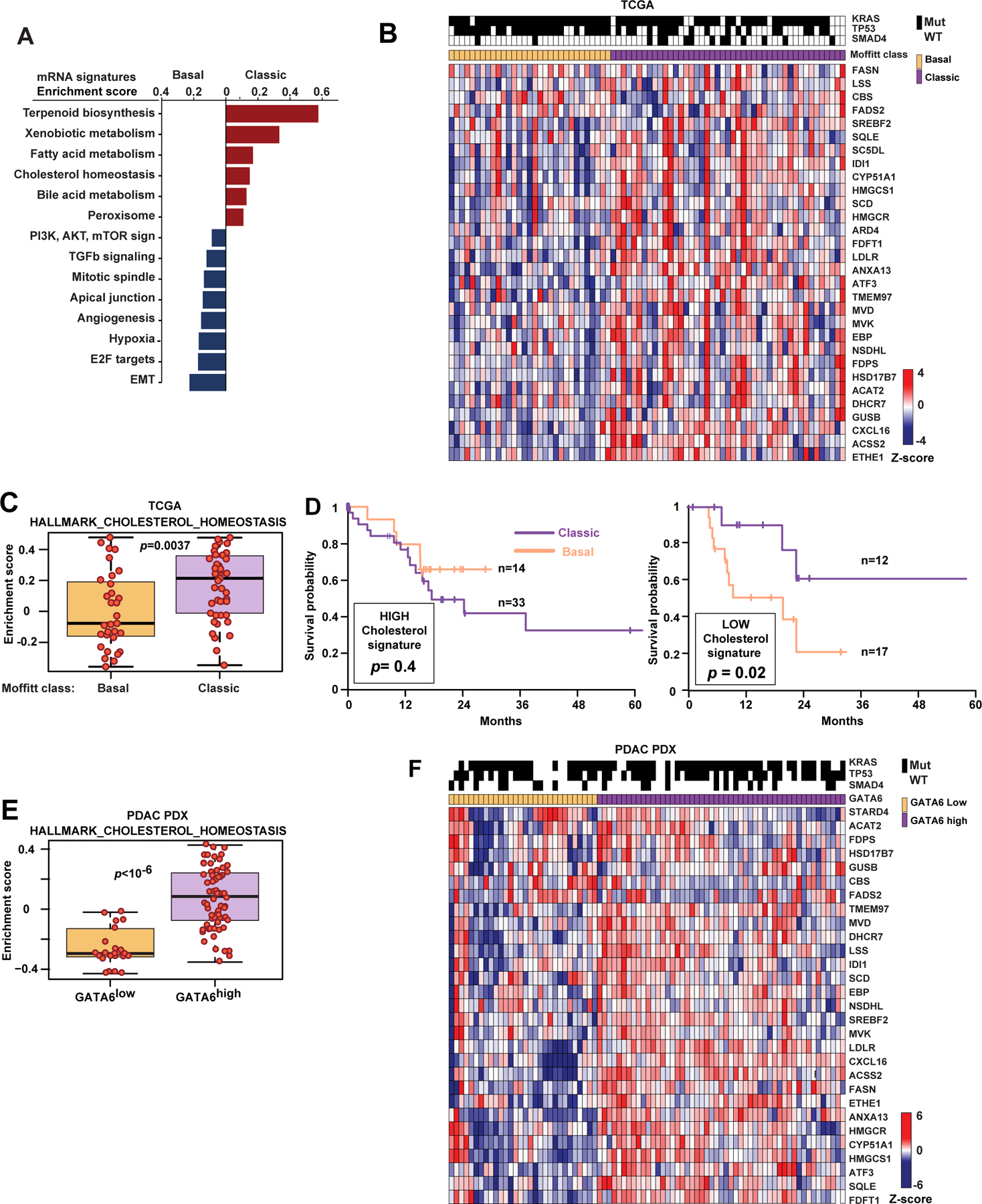
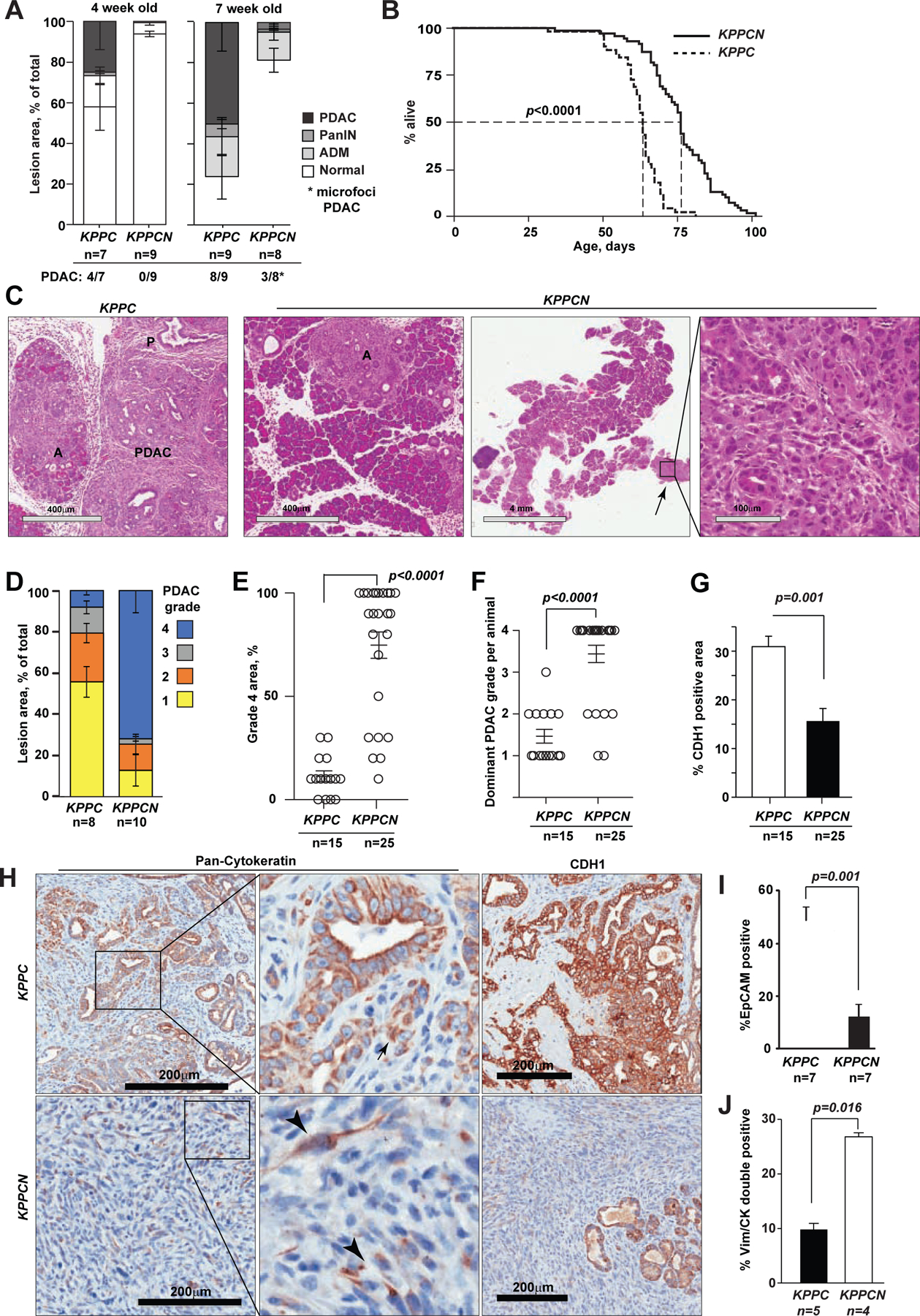
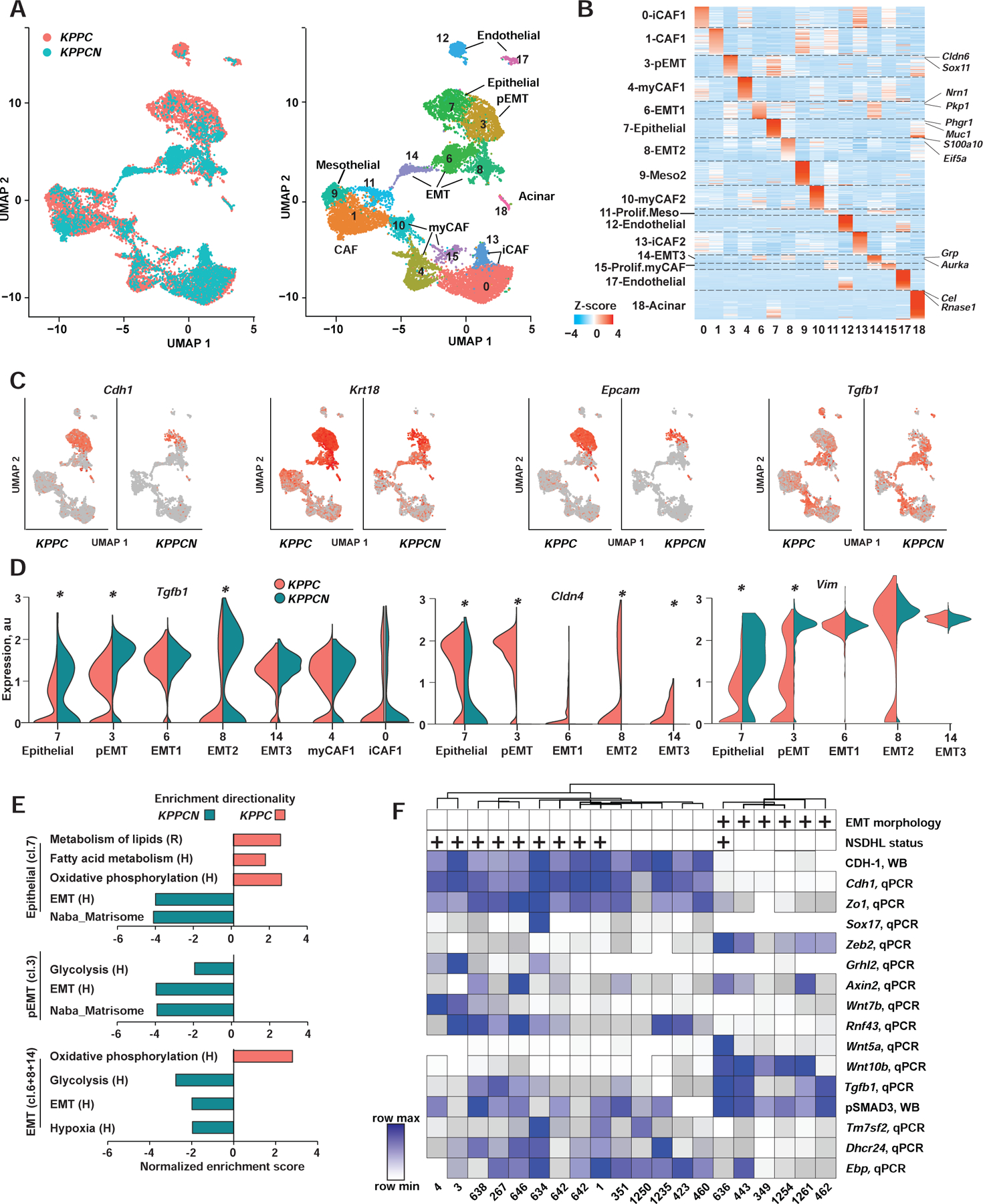
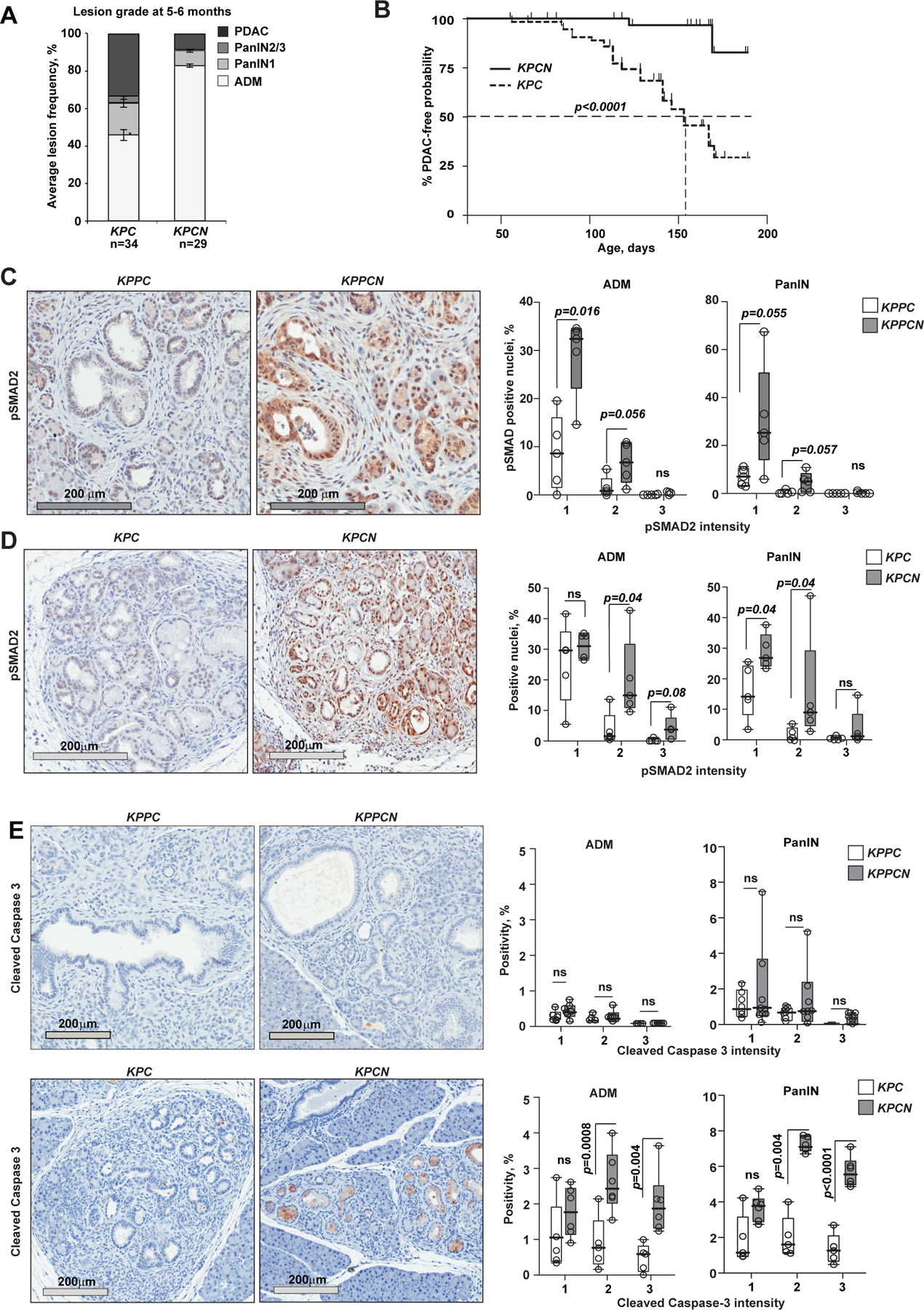
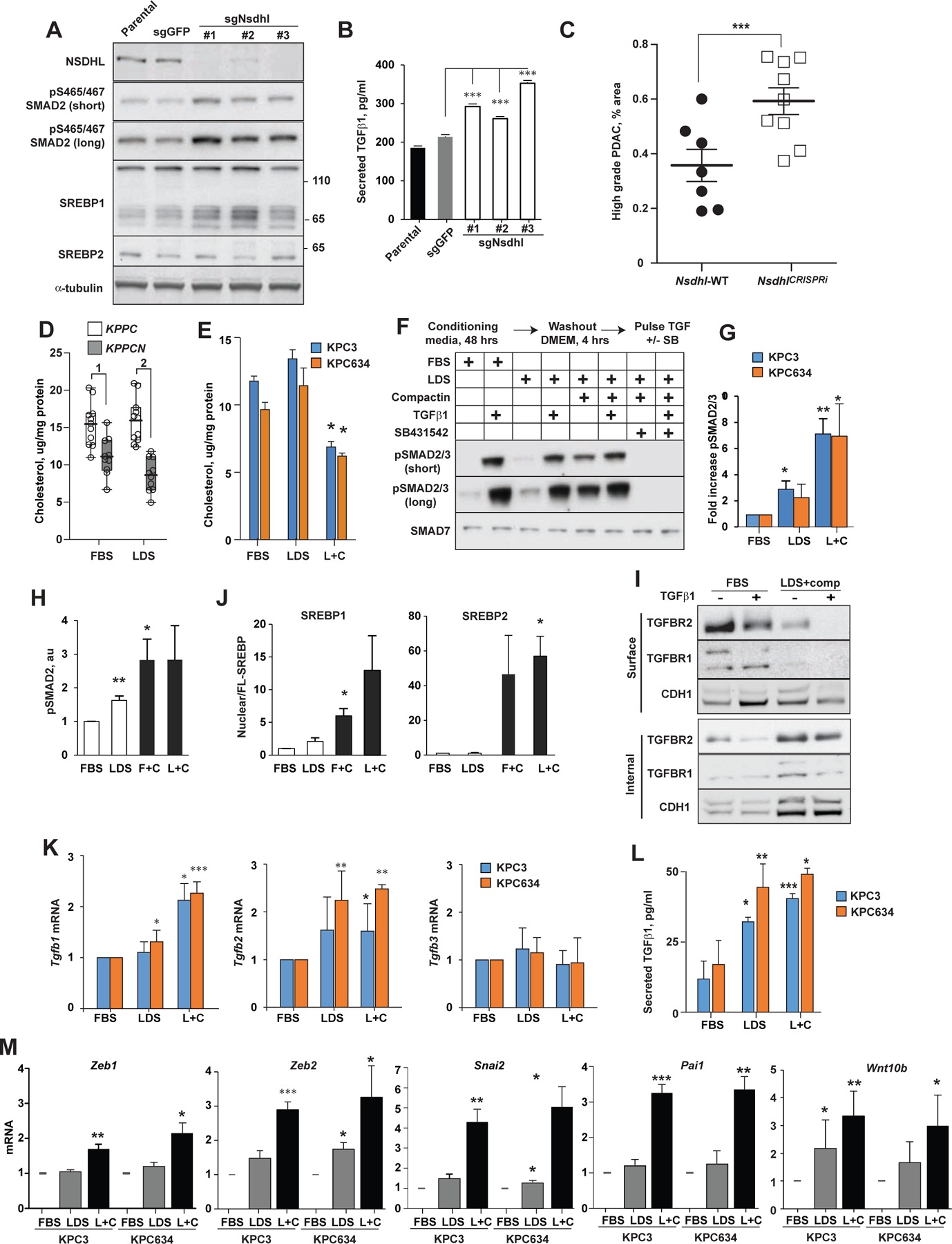
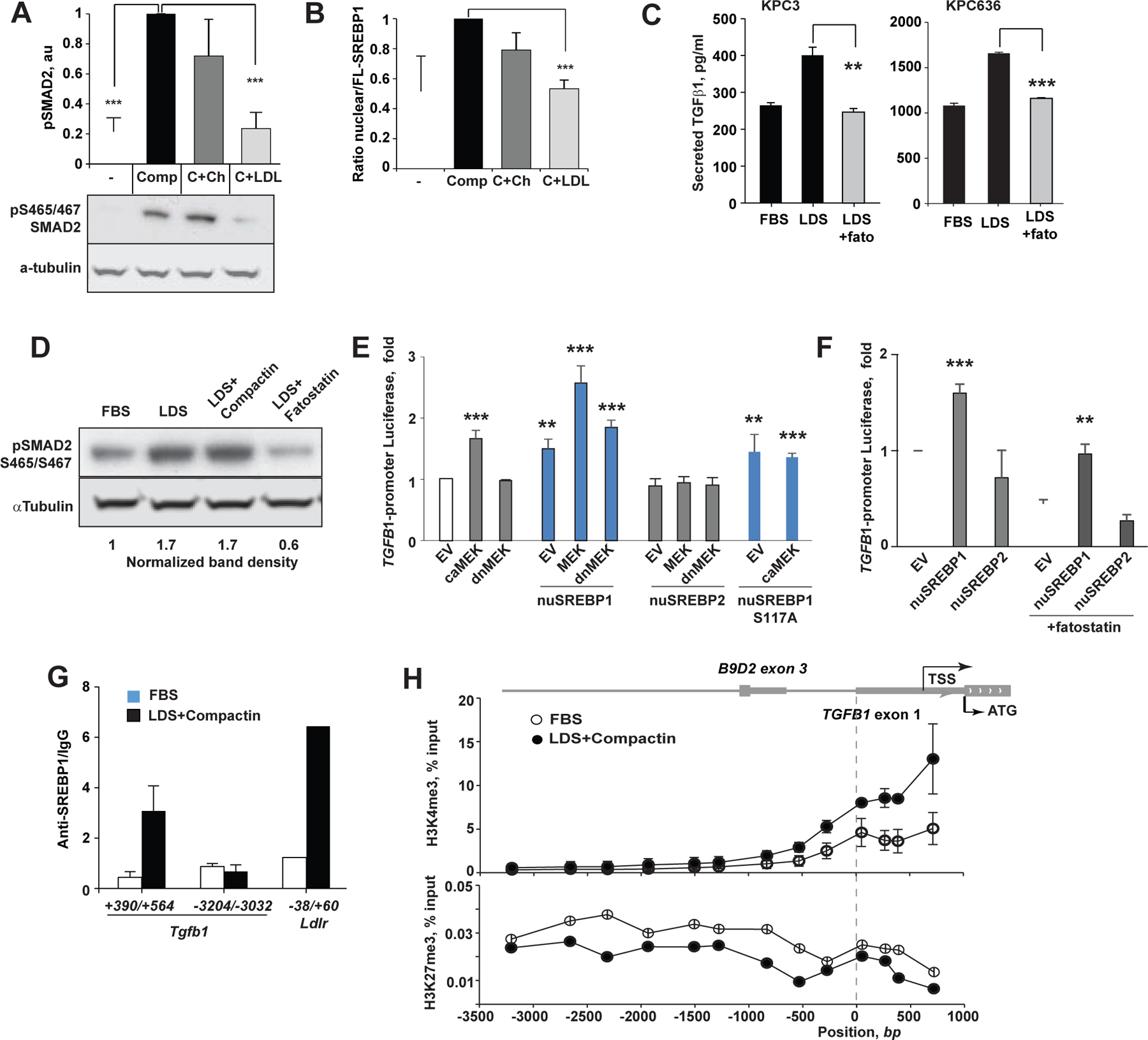
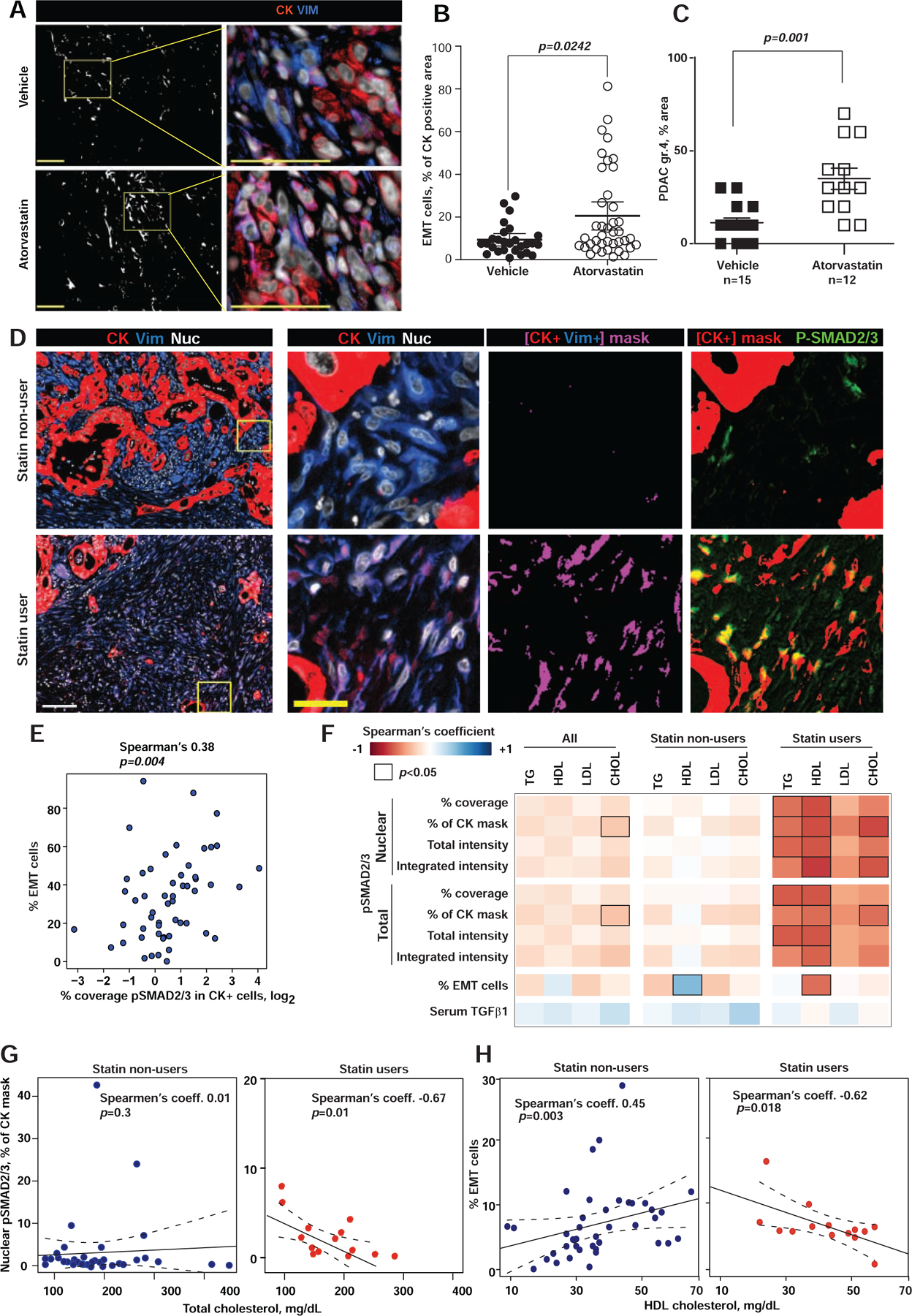
Oncogenic transformation alters lipid metabolism to sustain tumor growth. We define a mechanism by which cholesterol metabolism controls the development and differentiation of pancreatic ductal adenocarcinoma (PDAC). Disruption of distal cholesterol biosynthesis by conditional inactivation of the rate-limiting enzyme Nsdhl or treatment with cholesterol-lowering statins switches glandular pancreatic carcinomas to a basal (mesenchymal) phenotype in mouse models driven by KrasG12D expression and homozygous Trp53 loss. Consistently, PDACs in patients receiving statins show enhanced mesenchymal features. Mechanistically, statins and NSDHL loss induce SREBP1 activation, which promotes the expression of Tgfb1, enabling epithelial-mesenchymal transition. Evidence from patient samples in this study suggests that activation of transforming growth factor β signaling and epithelial-mesenchymal transition by cholesterol-lowering statins may promote the basal type of PDAC, conferring poor outcomes in patients.
Keywords: TGF-β signaling; cholesterol metabolism; epithelial-to-mesenchymal transition; pancreatic cancer.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests I.A. served as a consultant for Caris Life Sciences, Inc.
Figures
Comment in
-
Cholesterol Biosynthesis Influences Subtype Specificity and Plasticity in Pancreas Cancer.Cancer Cell. 2020 Oct 12;38(4):443-445. doi: 10.1016/j.ccell.2020.09.010. Cancer Cell. 2020. PMID: 33049206 Free PMC article.
References
-
- Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, and Goldstein JL (2004). Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. The Journal of biological chemistry 279, 52772–52780. - PubMed
-
- Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, Southwood B, Liang SB, Chadwick D, Zhang A, et al. (2018). Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 1344–1354. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R21 CA164205/CA/NCI NIH HHS/United States
- R21 CA231252/CA/NCI NIH HHS/United States
- S10 OD023666/OD/NIH HHS/United States
- R01 CA188430/CA/NCI NIH HHS/United States
- R50 CA221838/CA/NCI NIH HHS/United States
- K22 CA160725/CA/NCI NIH HHS/United States
- R01 CA232256/CA/NCI NIH HHS/United States
- R01 DK108195/DK/NIDDK NIH HHS/United States
- R01 HD065800/HD/NICHD NIH HHS/United States
- P30 CA006927/CA/NCI NIH HHS/United States
- T32 CA009035/CA/NCI NIH HHS/United States
- S10 OD023586/OD/NIH HHS/United States
- R01 CA113451/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
