

Genome-wide dynamics of RNA synthesis, processing, and degradation without RNA metabolic labeling
- PMID: 32978246
- PMCID: PMC7605262
- DOI: 10.1101/gr.260984.120
Genome-wide dynamics of RNA synthesis, processing, and degradation without RNA metabolic labeling
Abstract
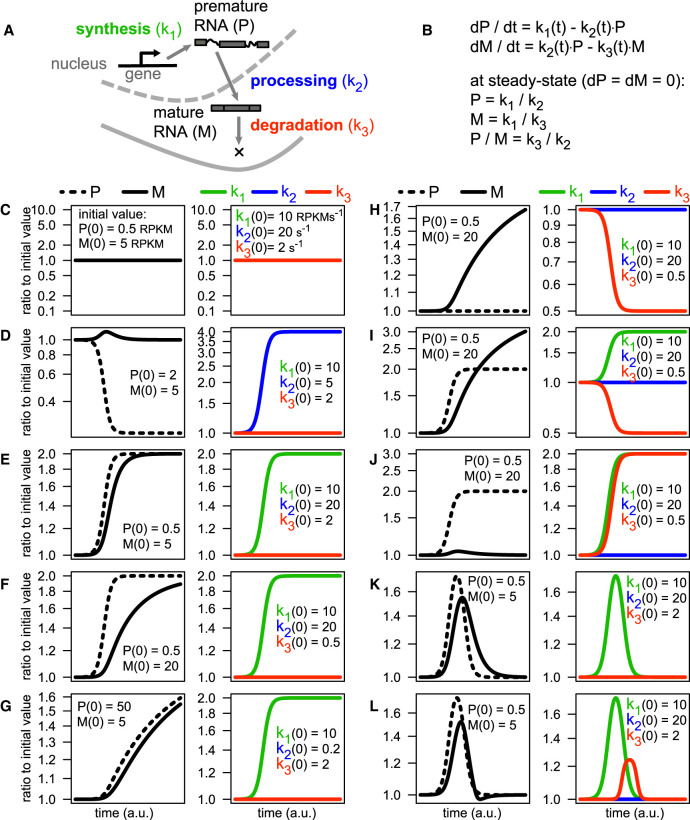
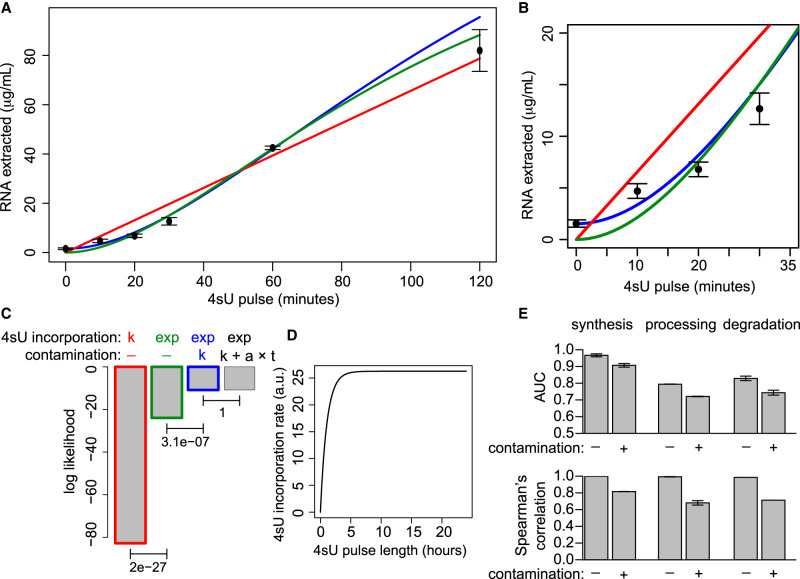
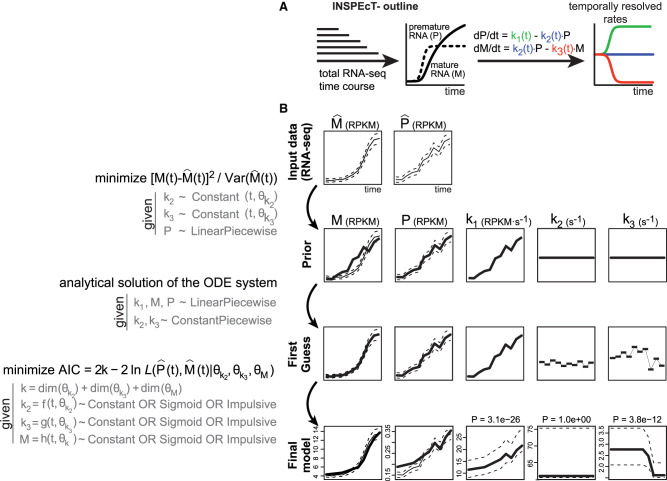
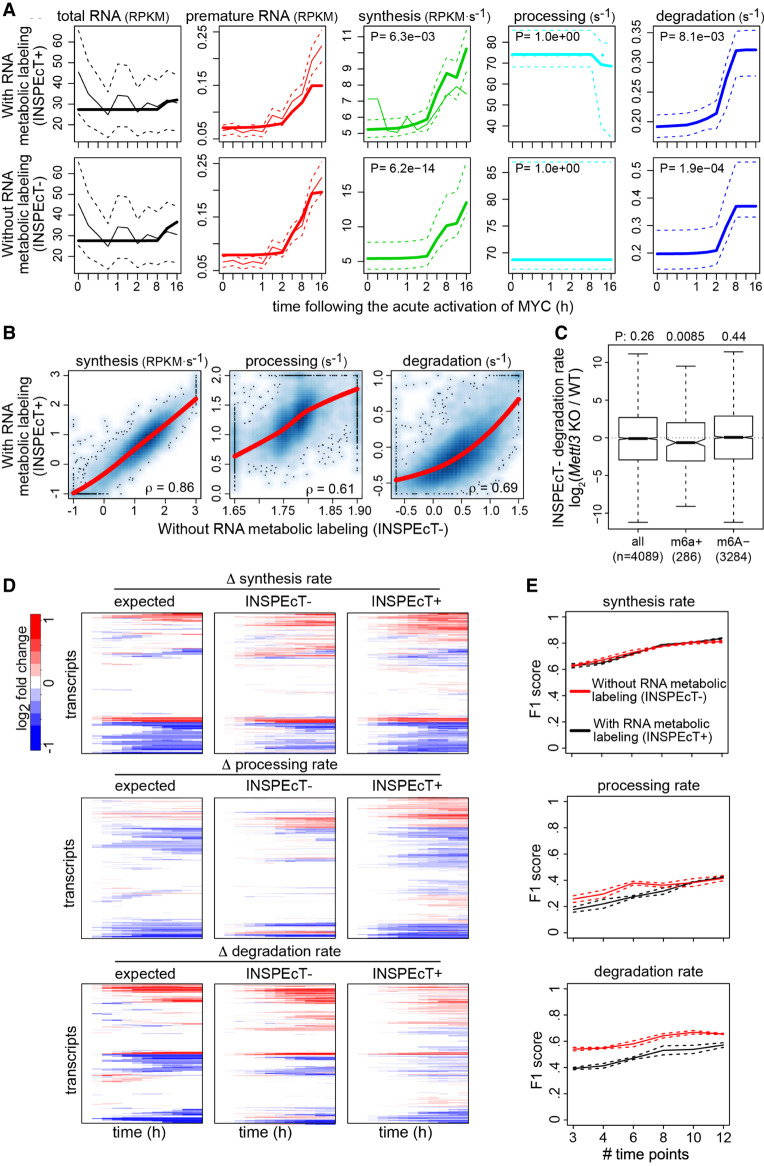
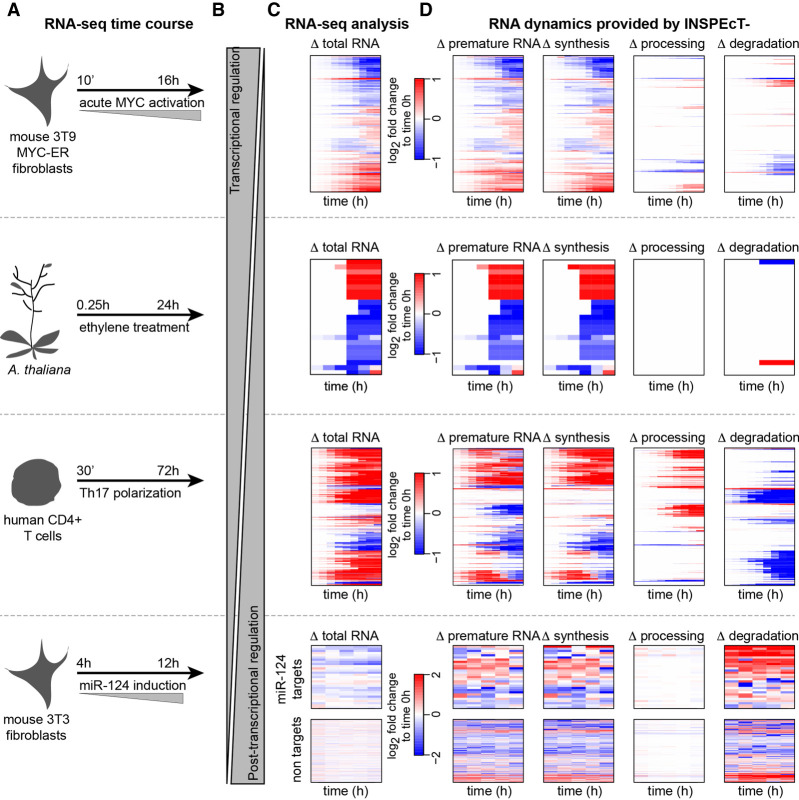
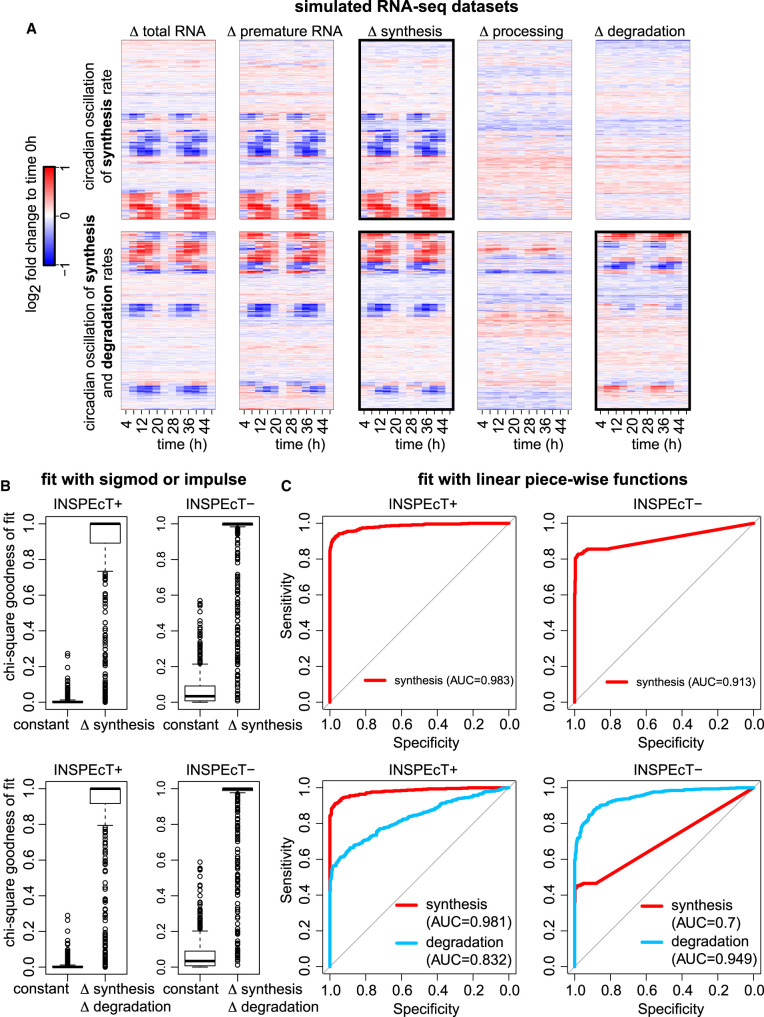
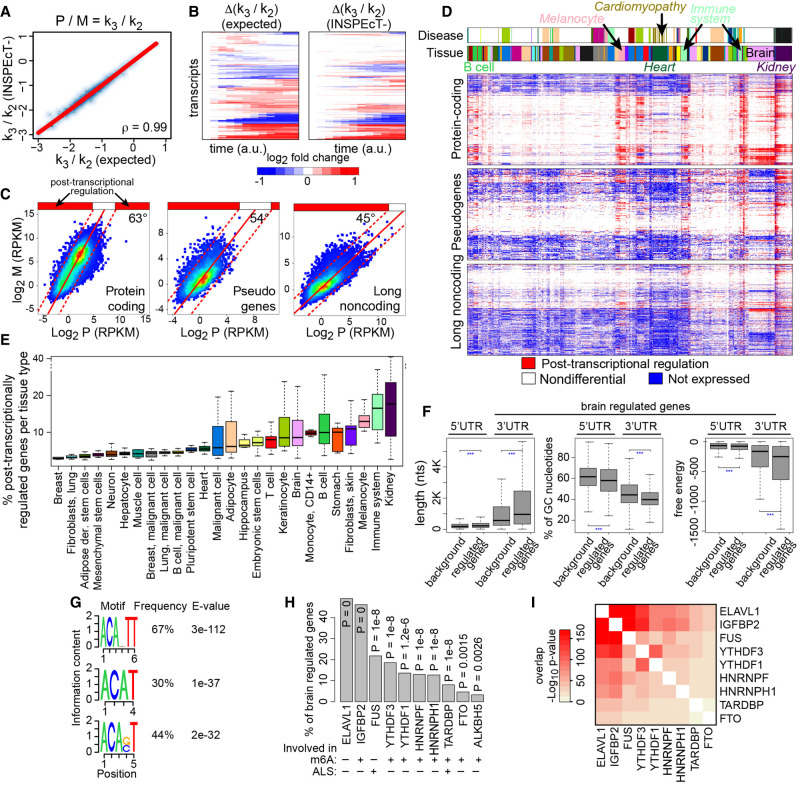
The quantification of the kinetic rates of RNA synthesis, processing, and degradation are largely based on the integrative analysis of total and nascent transcription, the latter being quantified through RNA metabolic labeling. We developed INSPEcT-, a computational method based on the mathematical modeling of premature and mature RNA expression that is able to quantify kinetic rates from steady-state or time course total RNA-seq data without requiring any information on nascent transcripts. Our approach outperforms available solutions, closely recapitulates the kinetic rates obtained through RNA metabolic labeling, improves the ability to detect changes in transcript half-lives, reduces the cost and complexity of the experiments, and can be adopted to study experimental conditions in which nascent transcription cannot be readily profiled. Finally, we applied INSPEcT- to the characterization of post-transcriptional regulation landscapes in dozens of physiological and disease conditions. This approach was included in the INSPEcT Bioconductor package, which can now unveil RNA dynamics from steady-state or time course data, with or without the profiling of nascent RNA.
© 2020 Furlan et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Austenaa LMI, Barozzi I, Simonatto M, Masella S, Chiara Della G, Ghisletti S, Curina A, de Wit E, Bouwman BAM, de Pretis S, et al. 2015. Transcription of mammalian cis-regulatory elements is restrained by actively enforced early termination. Mol Cell 60: 460–474. 10.1016/j.molcel.2015.09.018 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases