

Getting out what you put in: Copper in mitochondria and its impacts on human disease
- PMID: 32979421
- PMCID: PMC7680424
- DOI: 10.1016/j.bbamcr.2020.118867
Getting out what you put in: Copper in mitochondria and its impacts on human disease
Abstract
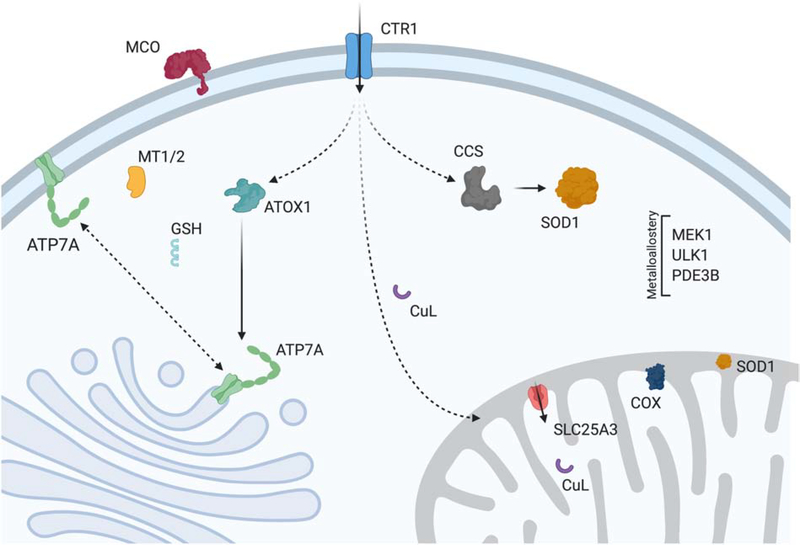
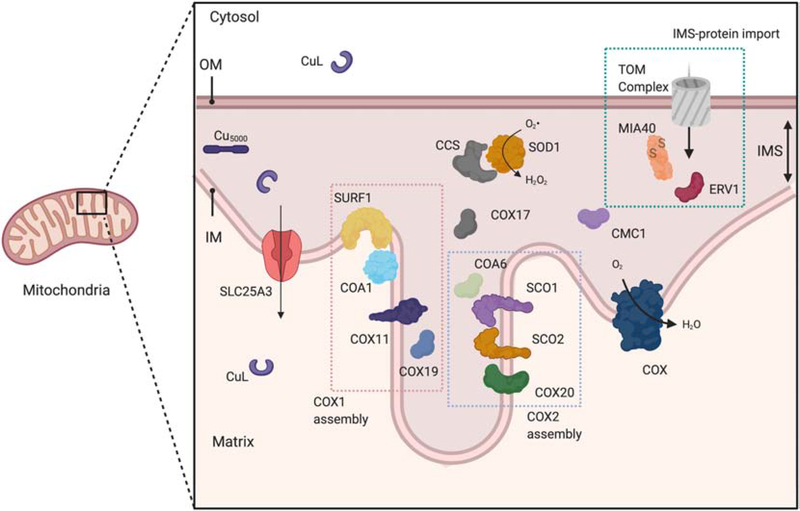
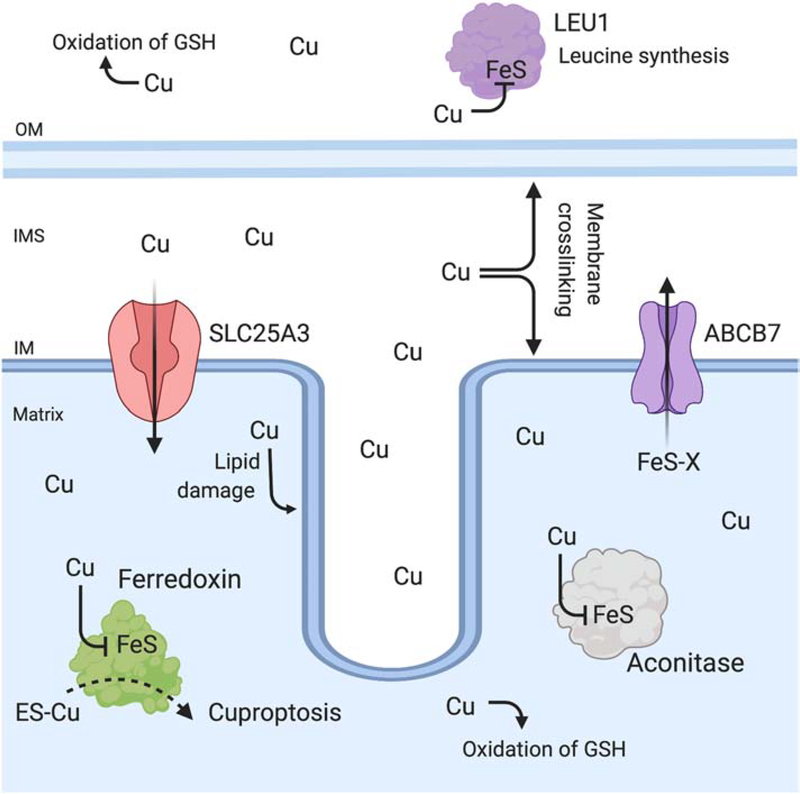
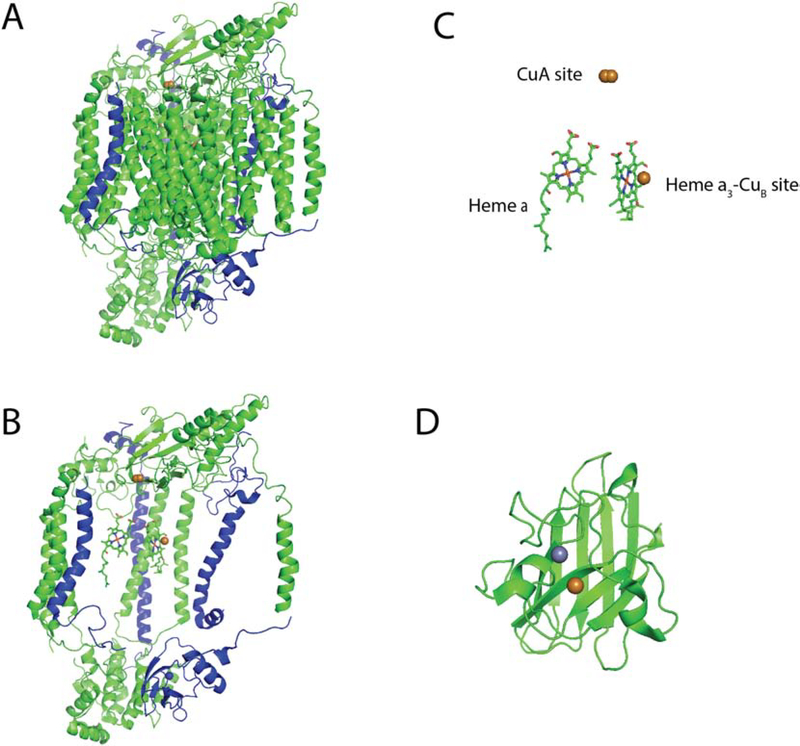
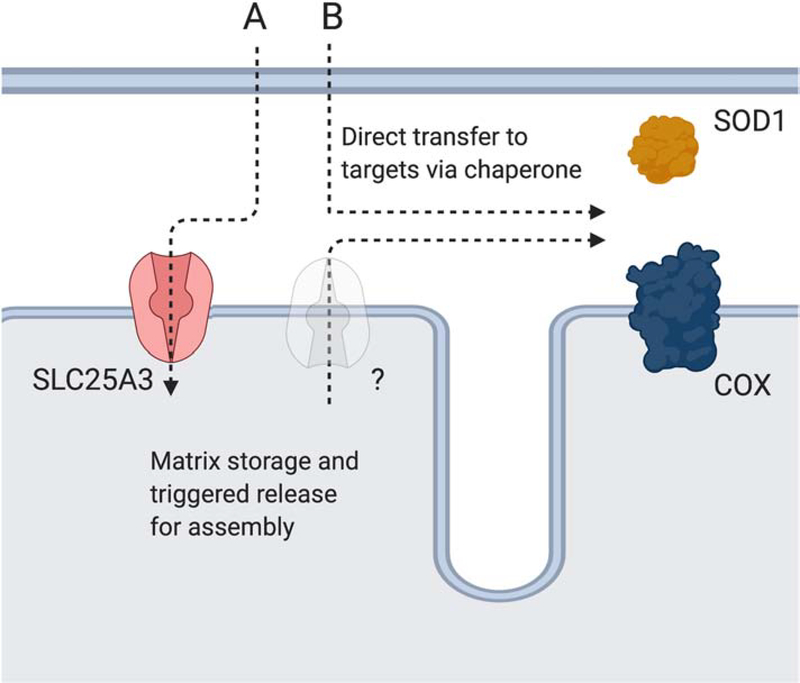
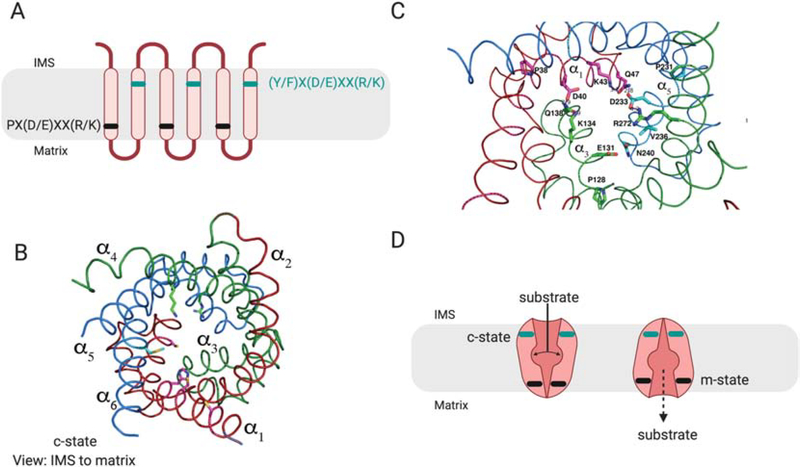
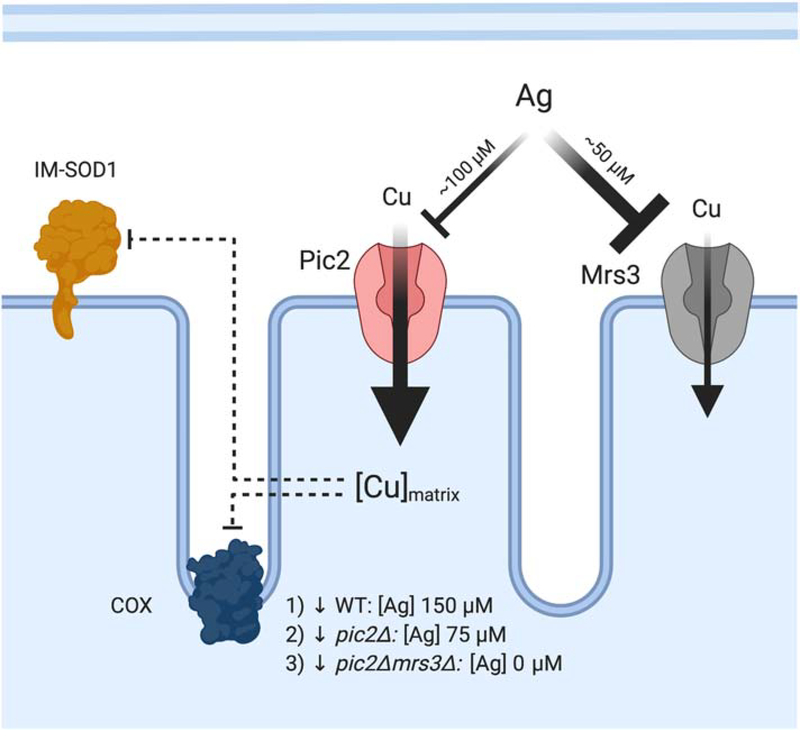
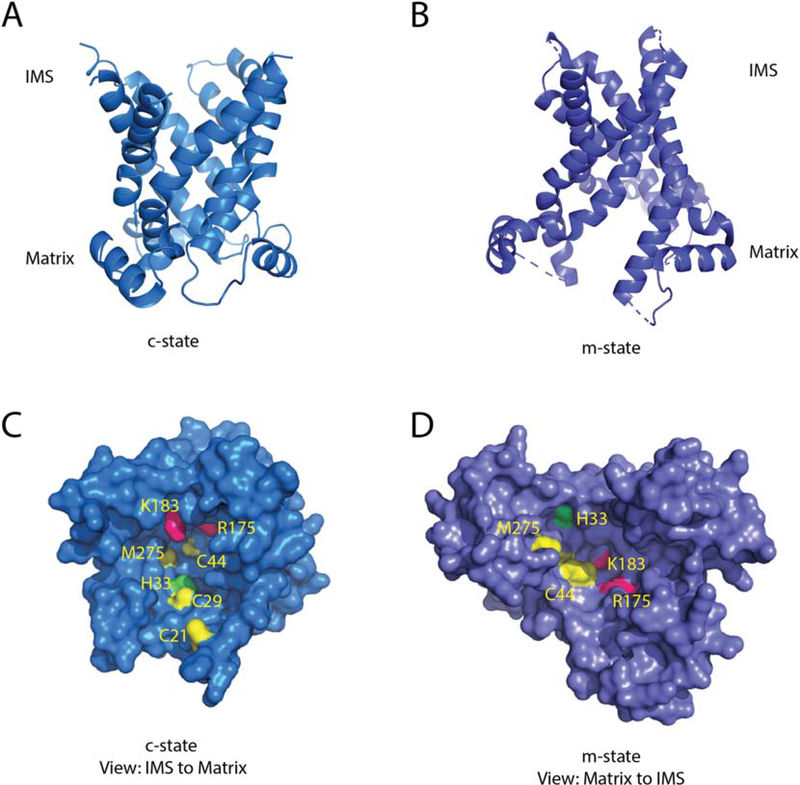
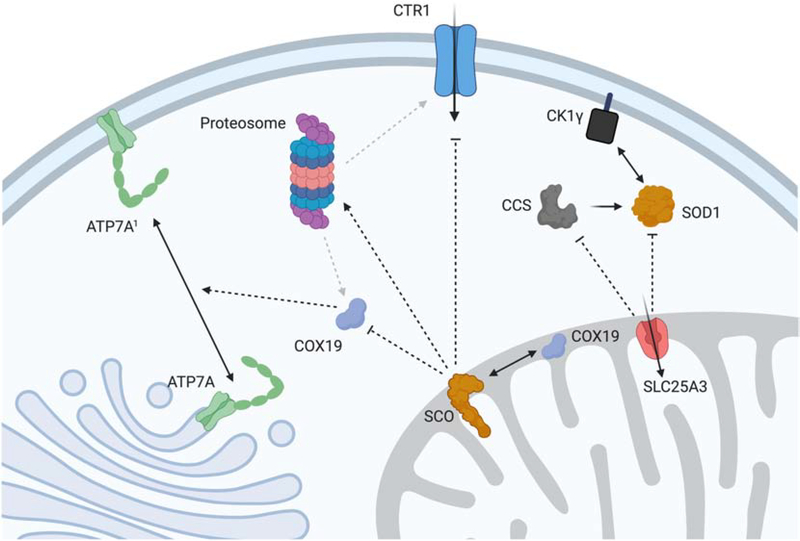
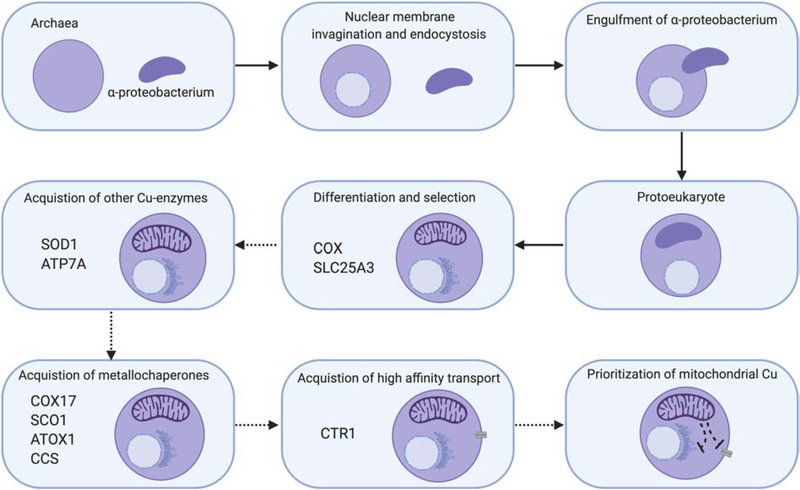
Mitochondria accumulate copper in their matrix for the eventual maturation of the cuproenzymes cytochrome c oxidase and superoxide dismutase. Transport into the matrix is achieved by mitochondrial carrier family (MCF) proteins. The major copper transporting MCF described to date in yeast is Pic2, which imports the metal ion into the matrix. Pic2 is one of ~30 MCFs that move numerous metabolites, nucleotides and co-factors across the inner membrane for use in the matrix. Genetic and biochemical experiments showed that Pic2 is required for cytochrome c oxidase activity under copper stress, and that it is capable of transporting ionic and complexed forms of copper. The Pic2 ortholog SLC25A3, one of 53 mammalian MCFs, functions as both a copper and a phosphate transporter. Depletion of SLC25A3 results in decreased accumulation of copper in the matrix, a cytochrome c oxidase defect and a modulation of cytosolic superoxide dismutase abundance. The regulatory roles for copper and cuproproteins resident to the mitochondrion continue to expand beyond the organelle. Mitochondrial copper chaperones have been linked to the modulation of cellular copper uptake and export and the facilitation of inter-organ communication. Recently, a role for matrix copper has also been proposed in a novel cell death pathway termed cuproptosis. This review will detail our understanding of the maturation of mitochondrial copper enzymes, the roles of mitochondrial signals in regulating cellular copper content, the proposed mechanisms of copper transport into the organelle and explore the evolutionary origins of copper homeostasis pathways.
Keywords: Copper; Cytochrome c oxidase; Mitochondria; Mitochondrial carrier family; Superoxide dismutase.
Copyright © 2020 Elsevier B.V. All rights reserved.
Figures
References
-
- Chang CJ, Searching for harmony in transition-metal signaling, Nat Chem Biol 11(10) (2015) 744–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
