

Efficient weighting methods for genomic best linear-unbiased prediction (BLUP) adapted to the genetic architectures of quantitative traits
- PMID: 32980863
- PMCID: PMC8027853
- DOI: 10.1038/s41437-020-00372-y
Efficient weighting methods for genomic best linear-unbiased prediction (BLUP) adapted to the genetic architectures of quantitative traits
Abstract
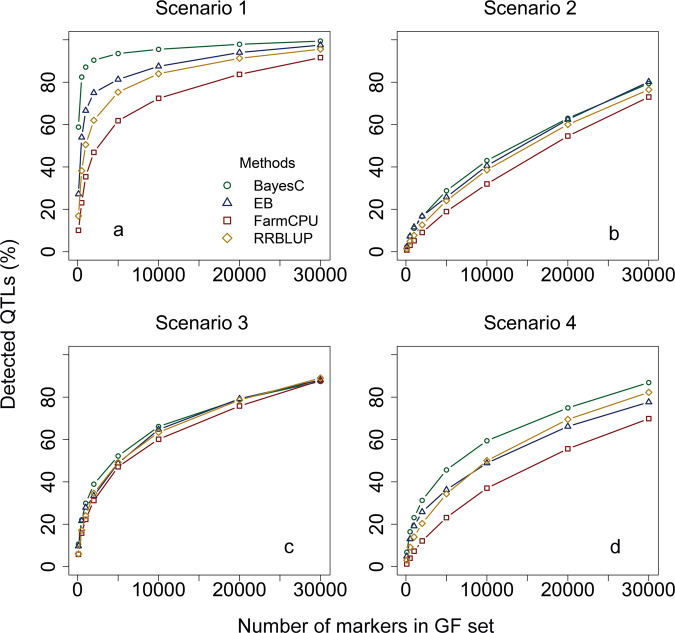
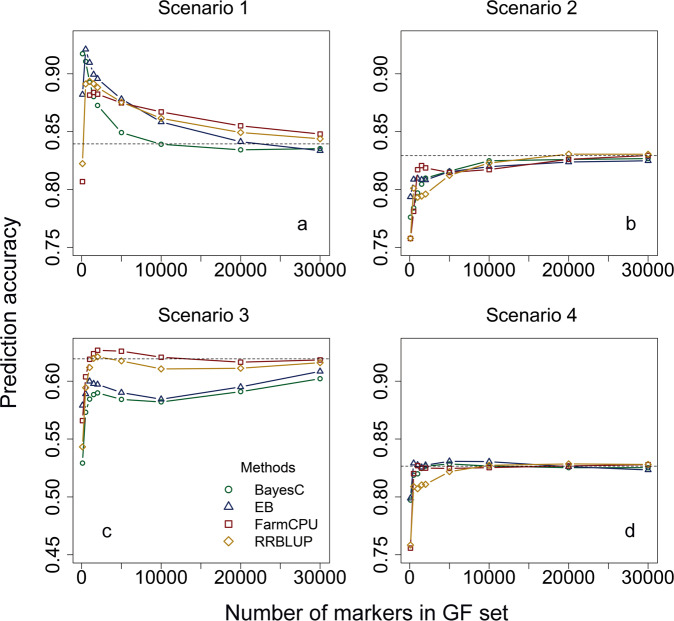
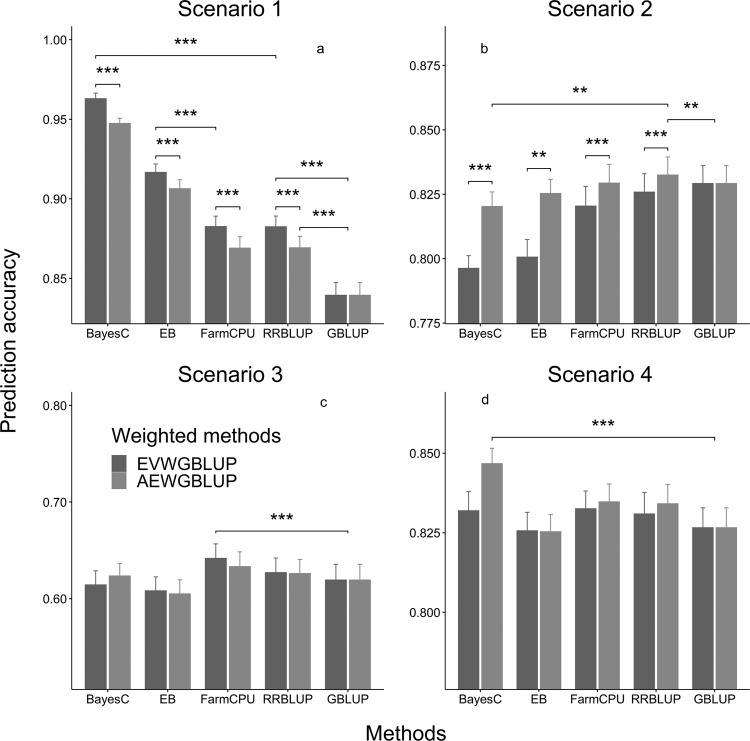
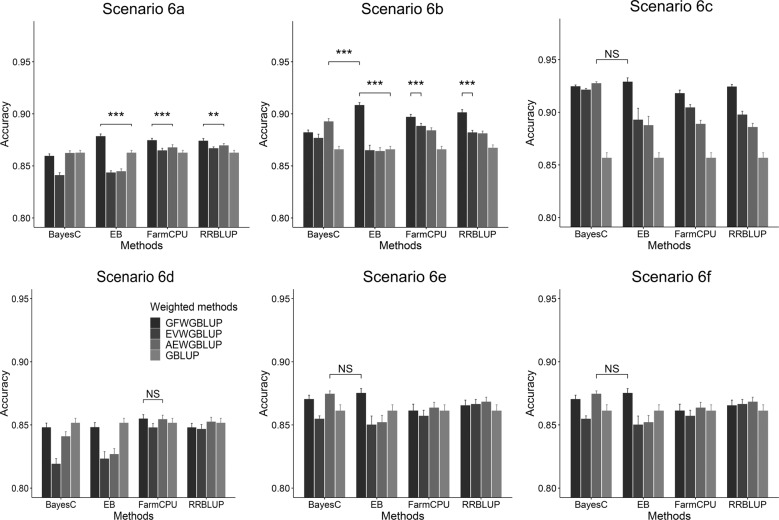
Genomic best linear-unbiased prediction (GBLUP) assumes equal variance for all marker effects, which is suitable for traits that conform to the infinitesimal model. For traits controlled by major genes, Bayesian methods with shrinkage priors or genome-wide association study (GWAS) methods can be used to identify causal variants effectively. The information from Bayesian/GWAS methods can be used to construct the weighted genomic relationship matrix (G). However, it remains unclear which methods perform best for traits varying in genetic architecture. Therefore, we developed several methods to optimize the performance of weighted GBLUP and compare them with other available methods using simulated and real data sets. First, two types of methods (marker effects with local shrinkage or normal prior) were used to obtain test statistics and estimates for each marker effect. Second, three weighted G matrices were constructed based on the marker information from the first step: (1) the genomic-feature-weighted G, (2) the estimated marker-variance-weighted G, and (3) the absolute value of the estimated marker-effect-weighted G. Following the above process, six different weighted GBLUP methods (local shrinkage/normal-prior GF/EV/AEWGBLUP) were proposed for genomic prediction. Analyses with both simulated and real data demonstrated that these options offer flexibility for optimizing the weighted GBLUP for traits with a broad spectrum of genetic architectures. The advantage of weighting methods over GBLUP in terms of accuracy was trait dependant, ranging from 14.8% to marginal for simulated traits and from 44% to marginal for real traits. Local-shrinkage prior EVWGBLUP is superior for traits mainly controlled by loci of a large effect. Normal-prior AEWGBLUP performs well for traits mainly controlled by loci of moderate effect. For traits controlled by some loci with large effects (explain 25-50% genetic variance) and a range of loci with small effects, GFWGBLUP has advantages. In conclusion, the optimal weighted GBLUP method for genomic selection should take both the genetic architecture and number of QTLs of traits into consideration carefully.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Performances of Adaptive MultiBLUP, Bayesian regressions, and weighted-GBLUP approaches for genomic predictions in Belgian Blue beef cattle.BMC Genomics. 2020 Aug 6;21(1):545. doi: 10.1186/s12864-020-06921-3. BMC Genomics. 2020. PMID: 32762654 Free PMC article.
-
Comparison of genomic predictions using genomic relationship matrices built with different weighting factors to account for locus-specific variances.J Dairy Sci. 2014 Oct;97(10):6547-59. doi: 10.3168/jds.2014-8210. Epub 2014 Aug 14. J Dairy Sci. 2014. PMID: 25129495
-
An efficient unified model for genome-wide association studies and genomic selection.Genet Sel Evol. 2017 Aug 24;49(1):64. doi: 10.1186/s12711-017-0338-x. Genet Sel Evol. 2017. PMID: 28836943 Free PMC article.
-
Application of Bayesian genomic prediction methods to genome-wide association analyses.Genet Sel Evol. 2022 May 13;54(1):31. doi: 10.1186/s12711-022-00724-8. Genet Sel Evol. 2022. PMID: 35562659 Free PMC article. Review.
-
Accounting for Correlation Between Traits in Genomic Prediction.Methods Mol Biol. 2022;2467:285-327. doi: 10.1007/978-1-0716-2205-6_10. Methods Mol Biol. 2022. PMID: 35451780 Review.
Cited by
-
Improvement of Genomic Predictions in Small Breeds by Construction of Genomic Relationship Matrix Through Variable Selection.Front Genet. 2022 May 18;13:814264. doi: 10.3389/fgene.2022.814264. eCollection 2022. Front Genet. 2022. PMID: 35664297 Free PMC article.
-
deepGBLUP: joint deep learning networks and GBLUP framework for accurate genomic prediction of complex traits in Korean native cattle.Genet Sel Evol. 2023 Jul 31;55(1):56. doi: 10.1186/s12711-023-00825-y. Genet Sel Evol. 2023. PMID: 37525091 Free PMC article.
-
Benchmarking machine learning and parametric methods for genomic prediction of feed efficiency-related traits in Nellore cattle.Sci Rep. 2024 Mar 17;14(1):6404. doi: 10.1038/s41598-024-57234-4. Sci Rep. 2024. PMID: 38493207 Free PMC article.
-
Preselecting Variants from Large-Scale Genome-Wide Association Study Meta-Analyses Increases the Genomic Prediction Accuracy of Growth and Carcass Traits in Large White Pigs.Animals (Basel). 2023 Dec 5;13(24):3746. doi: 10.3390/ani13243746. Animals (Basel). 2023. PMID: 38136785 Free PMC article.
-
PNNGS, a multi-convolutional parallel neural network for genomic selection.Front Plant Sci. 2024 Sep 3;15:1410596. doi: 10.3389/fpls.2024.1410596. eCollection 2024. Front Plant Sci. 2024. PMID: 39290743 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
