

Human Autoinflammatory Diseases Mediated by NLRP3-, Pyrin-, NLRP1-, and NLRC4-Inflammasome Dysregulation Updates on Diagnosis, Treatment, and the Respective Roles of IL-1 and IL-18
- PMID: 32983099
- PMCID: PMC7477077
- DOI: 10.3389/fimmu.2020.01840
Human Autoinflammatory Diseases Mediated by NLRP3-, Pyrin-, NLRP1-, and NLRC4-Inflammasome Dysregulation Updates on Diagnosis, Treatment, and the Respective Roles of IL-1 and IL-18
Abstract
Recent research has led to novel findings in inflammasome biology and genetics that altered the diagnosis and management of patients with autoinflammatory syndromes caused by NLRP3-, Pyrin-, NLRP1-, and NLRC4-inflammasomes and spurred the development of novel treatments. The use of next-generation sequencing in clinical practice allows for rapid diagnosis and the detection of somatic mutations that cause autoinflammatory diseases. Clinical differences in patients with NLRP3, pyrin, and NLRP1 inflammasomopathies, and the constitutive elevation of unbound free serum IL-18 that predisposes to the development of macrophage activation syndrome (MAS) in patients with gain-of function mutations in NLRC4 led to the screening and the characterization of novel diseases presenting with constitutively elevated serum IL-18 levels, and start to unravel the biology of "high IL-18 states" that translate into the use of biomarkers that improve diagnosis and monitoring of disease activity and investigations of treatments that target IL-18 and IFN-gamma which promise to improve the management and outcome of these conditions. Lastly, advances in structural modeling by cryo-electron microscopy (cryo-EM) of gasdermin, and of NLRP3- and NLRC4-inflammasome assembly, and the characterization of post-translational modifications (PTM) that regulate inflammasome activation, coupled with high-throughput screening (HTS) of libraries of inflammasome-inhibiting compounds, promise a new generation of treatments for patients with inflammasome-mediated diseases.
Keywords: GSDMD; NLRC4; NLRP1; NLRP3; autoinflammatory diseases; inflammasome; pyrin.
Copyright © 2020 Alehashemi and Goldbach-Mansky.
Figures

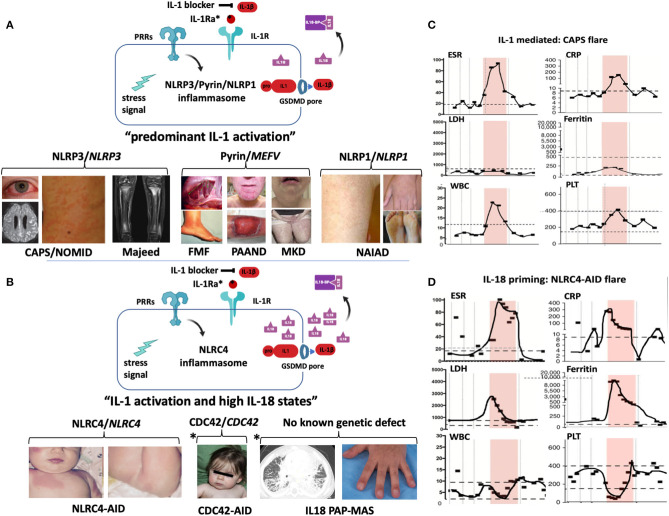
 ) that cause Majeed syndrome (depicted is sterile osteomyelitis of the growth plates, the disease is not discussed in the text). Additive GOF mutations in pyrin cause familial Mediterranean fever (FMF) (depicted are abdominal adhesions that can develop in chronic sterile peritonitis and erysipelas-like erythema of the ankle in FMF) or Pyrin-Associated Autoinflammatory with Neutrophilic Dermatosis (PAAND) (depicted is cystic acne and pyoderma gangrenosum in a patient with neutrophilic dermatitis), and secondary pyrin inflammasome activation is caused by LOF mutations in MVK that cause Mevalonate kinase deficiency (MKD) (depicted is lymphadenitis and a papular rash). GOF mutations in NLRP1 cause NLRP1-Associated Autoinflammation with Arthritis and Dyskeratosis (NAIAD) (depicted is follicular dyskeratosis, hyperkeratosis of the soles and lesions on left hand). (B) GOF mutations in NLRC4 cause the NLRC4-Associated Autoinflammatory Disorders (NLRC4-AID) associated with often ultra-high serum IL-18 expression which predisposes to the development of MAS [images depict the rash from a patient with NLRC4-associated macrophage activation syndrome (MAS)]. Other high IL-18 states include CDC42 mediated autoinflammatory disease (depicted are mild facial dysmorphisms including frontal bossing and nasal bridge depression); IL-18 PAP-MAS (a subset of SOJIA-ILD), an autoinflammatory disease without known genetic defect (depicted are a chest CT scan with interstitial lung disease and clubbing of the fingernails). For the 2 latter diseases, increased NLRC4 inflammasome activation as cause of the high IL-18 has not been demonstrated. (A,B) Inflammatory markers during MAS and CAPS flares differ. Flare episodes in patients with CAPS (C) differ from flare episodes in patients with high IL-18 levels (shown in a patient with NLRC4-AID in D). ESR and CRP are similarly elevated in both diseases, in MAS other features include elevated ferritin and LDH, and cytopenias (leukopenia, thrombocytopenia); while in CAPS, ferritin and LDH increase little if at all and patients develop leukocytosis and thrombocytosis. (The shaded area shows longitudinal laboratory markers collected during a 2-week period during a disease flare). CAPS, Cryopyrin-Associated Periodic Syndrome; CDC42, Cell Division Control protein 42 homolog; FMF, Familial Mediterranean Fever; IL18-BP, IL-18 binding protein; IL-1Ra, IL-1 receptor antagonist; MKD or HIDS, Mevalonate Kinase Deficiency or Hyper-IgD Syndrome; NAIAD, NLRP1 Associated Autoinflammation with Arthritis and Dyskeratosis; NLRC4-AID, NLRC4-Associated Autoinflammatory Disorders; NOMID, Neonatal-Onset Multisystem Inflammatory Disease; PAAND, Pyrin-Associated Autoinflammatory with Neutrophilic Dermatosis; PAP-MAS, Pulmonary Alveolar Proteinosis-Macrophage Activation Syndrome; PRR, Pattern recognition receptor.
) that cause Majeed syndrome (depicted is sterile osteomyelitis of the growth plates, the disease is not discussed in the text). Additive GOF mutations in pyrin cause familial Mediterranean fever (FMF) (depicted are abdominal adhesions that can develop in chronic sterile peritonitis and erysipelas-like erythema of the ankle in FMF) or Pyrin-Associated Autoinflammatory with Neutrophilic Dermatosis (PAAND) (depicted is cystic acne and pyoderma gangrenosum in a patient with neutrophilic dermatitis), and secondary pyrin inflammasome activation is caused by LOF mutations in MVK that cause Mevalonate kinase deficiency (MKD) (depicted is lymphadenitis and a papular rash). GOF mutations in NLRP1 cause NLRP1-Associated Autoinflammation with Arthritis and Dyskeratosis (NAIAD) (depicted is follicular dyskeratosis, hyperkeratosis of the soles and lesions on left hand). (B) GOF mutations in NLRC4 cause the NLRC4-Associated Autoinflammatory Disorders (NLRC4-AID) associated with often ultra-high serum IL-18 expression which predisposes to the development of MAS [images depict the rash from a patient with NLRC4-associated macrophage activation syndrome (MAS)]. Other high IL-18 states include CDC42 mediated autoinflammatory disease (depicted are mild facial dysmorphisms including frontal bossing and nasal bridge depression); IL-18 PAP-MAS (a subset of SOJIA-ILD), an autoinflammatory disease without known genetic defect (depicted are a chest CT scan with interstitial lung disease and clubbing of the fingernails). For the 2 latter diseases, increased NLRC4 inflammasome activation as cause of the high IL-18 has not been demonstrated. (A,B) Inflammatory markers during MAS and CAPS flares differ. Flare episodes in patients with CAPS (C) differ from flare episodes in patients with high IL-18 levels (shown in a patient with NLRC4-AID in D). ESR and CRP are similarly elevated in both diseases, in MAS other features include elevated ferritin and LDH, and cytopenias (leukopenia, thrombocytopenia); while in CAPS, ferritin and LDH increase little if at all and patients develop leukocytosis and thrombocytosis. (The shaded area shows longitudinal laboratory markers collected during a 2-week period during a disease flare). CAPS, Cryopyrin-Associated Periodic Syndrome; CDC42, Cell Division Control protein 42 homolog; FMF, Familial Mediterranean Fever; IL18-BP, IL-18 binding protein; IL-1Ra, IL-1 receptor antagonist; MKD or HIDS, Mevalonate Kinase Deficiency or Hyper-IgD Syndrome; NAIAD, NLRP1 Associated Autoinflammation with Arthritis and Dyskeratosis; NLRC4-AID, NLRC4-Associated Autoinflammatory Disorders; NOMID, Neonatal-Onset Multisystem Inflammatory Disease; PAAND, Pyrin-Associated Autoinflammatory with Neutrophilic Dermatosis; PAP-MAS, Pulmonary Alveolar Proteinosis-Macrophage Activation Syndrome; PRR, Pattern recognition receptor.References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous