

16S Metagenomics Reveals Dysbiosis of Nasal Core Microbiota in Children With Chronic Nasal Inflammation: Role of Adenoid Hypertrophy and Allergic Rhinitis
- PMID: 32984078
- PMCID: PMC7492700
- DOI: 10.3389/fcimb.2020.00458
16S Metagenomics Reveals Dysbiosis of Nasal Core Microbiota in Children With Chronic Nasal Inflammation: Role of Adenoid Hypertrophy and Allergic Rhinitis
Abstract
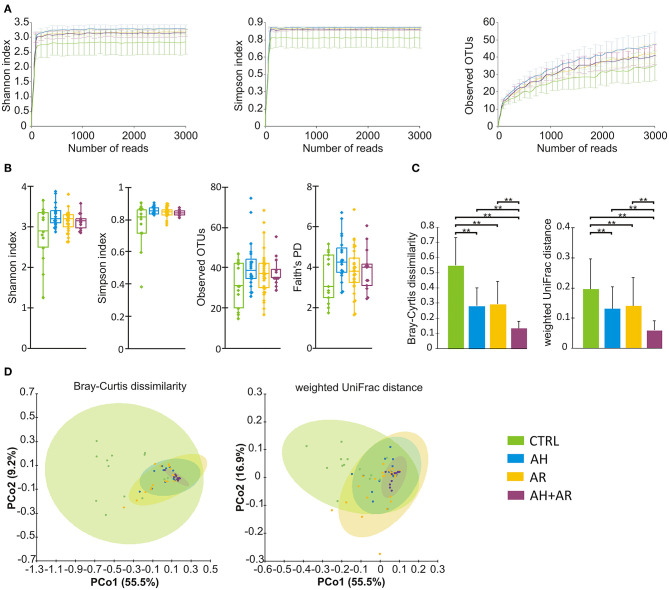
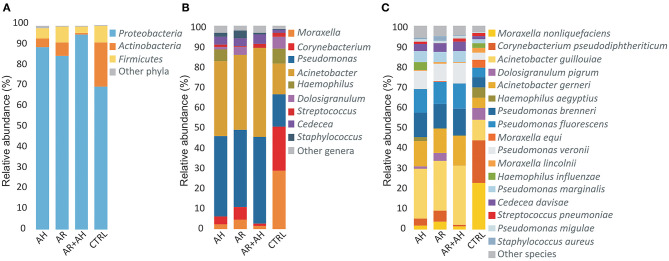
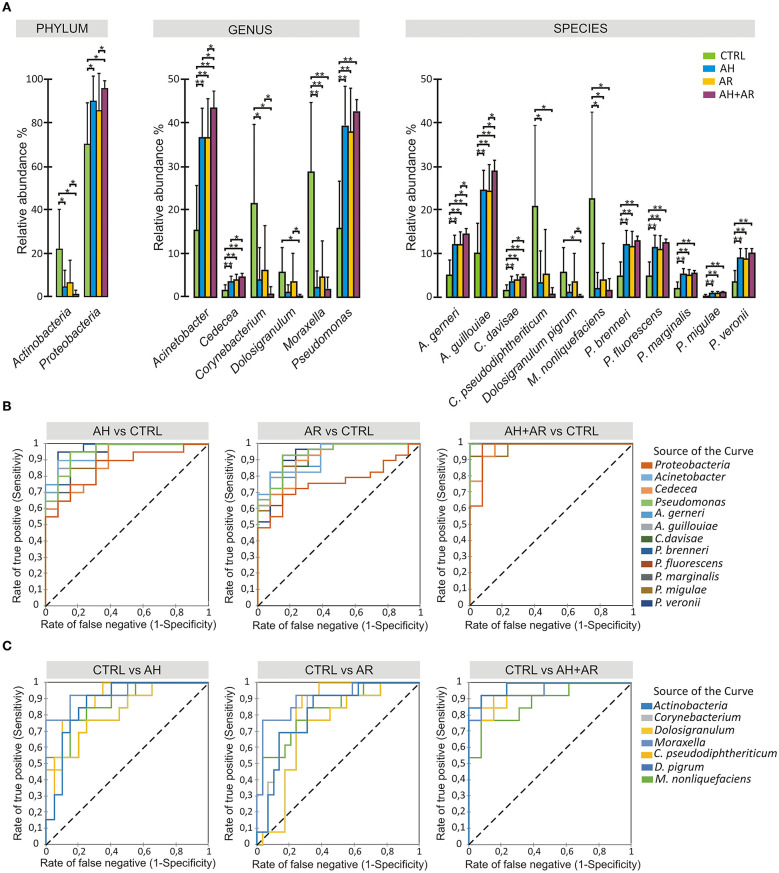
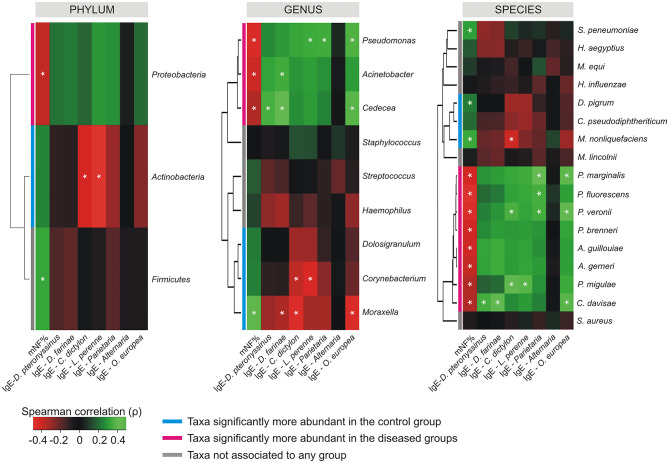
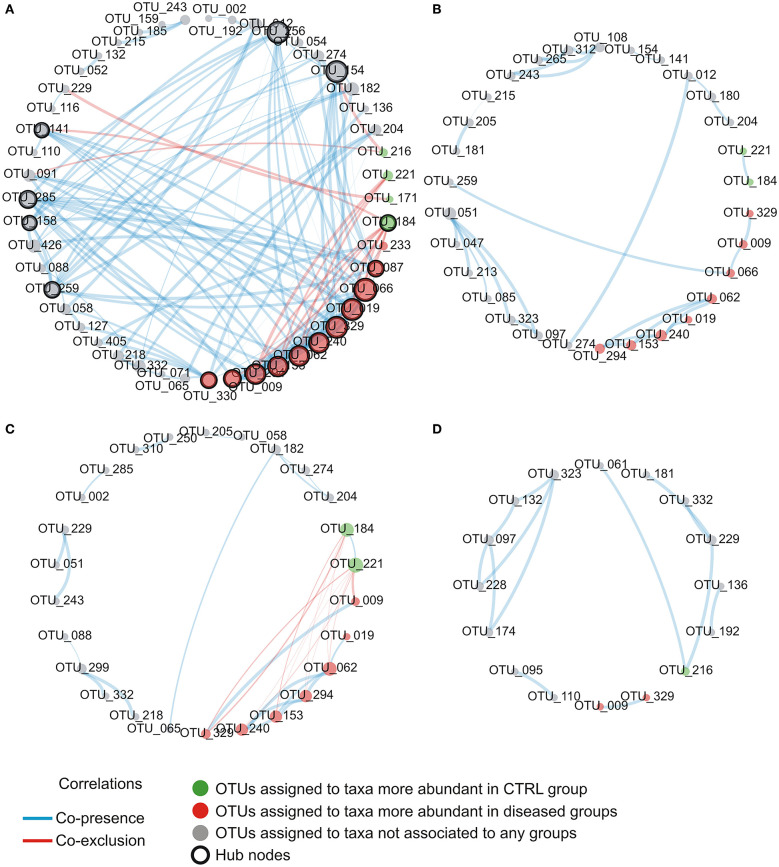
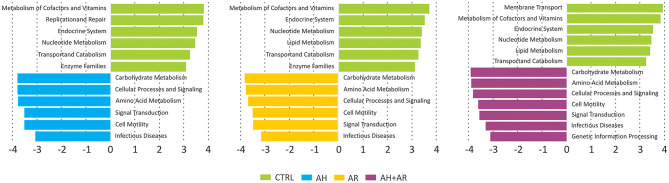
Allergic rhinitis (AR) and adenoid hypertrophy (AH) are, in children, the main cause of partial or complete upper airway obstruction and reduction in airflow. However, limited data exist about the impact of the increased resistance to airflow, on the nasal microbial composition of children with AR end AH. Allergic rhinitis (AR) as well as adenoid hypertrophy (AH), represent extremely common pathologies in this population. Their known inflammatory obstruction is amplified when both pathologies coexist. In our study, the microbiota of anterior nares of 75 pediatric subjects with AR, AH or both conditions, was explored by 16S rRNA-based metagenomic approach. Our data show for the first time, that in children, the inflammatory state is associated to similar changes in the microbiota composition of AR and AH subjects respect to the healthy condition. Together with such alterations, we observed a reduced variability in the between-subject biodiversity on the other hand, these same alterations resulted amplified by the nasal obstruction that could constitute a secondary risk factor for dysbiosis. Significant differences in the relative abundance of specific microbial groups were found between diseased phenotypes and the controls. Most of these taxa belonged to a stable and quantitatively dominating component of the nasal microbiota and showed marked potentials in discriminating the controls from diseased subjects. A pauperization of the nasal microbial network was observed in diseased status in respect to the number of involved taxa and connectivity. Finally, while stable co-occurrence relationships were observed within both control- and diseases-associated microbial groups, only negative correlations were present between them, suggesting that microbial subgroups potentially act as maintainer of the eubiosis state in the nasal ecosystem. In the nasal ecosystem, inflammation-associated shifts seem to impact the more intimate component of the microbiota rather than representing the mere loss of microbial diversity. The discriminatory potential showed by differentially abundant taxa provide a starting point for future research with the potential to improve patient outcomes. Overall, our results underline the association of AH and AR with the impairment of the microbial interplay leading to unbalanced ecosystems.
Keywords: adenoid hypertrophy; allergic rhinitis; chronic inflammation; core microbiota; nasal microbiota.
Copyright © 2020 Marazzato, Zicari, Aleandri, Conte, Longhi, Vitanza, Bolognino, Zagaglia, De Castro, Brindisi, Schiavi, De Vittori, Reddel, Quagliariello, Del Chierico, Putignani, Duse, Palamara and Conte.
Figures
References
-
- Biesbroek G., Tsivtsivadze E., Sanders E. A., Montijn R., Veenhoven R. H., Keijser B. J. F., et al. . (2014). Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children. Am. J. Respir. Crit. Care Med. 190, 1283–1292. 10.1164/rccm.201407-1240OC - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials