

Gene Therapy of the Hemoglobinopathies
- PMID: 32984772
- PMCID: PMC7489710
- DOI: 10.1097/HS9.0000000000000479
Gene Therapy of the Hemoglobinopathies
Abstract
Sickle cell disease and the ß-thalassemias are caused by mutations of the ß-globin gene and represent the most frequent single gene disorders worldwide. Even in European countries with a previous low frequency of these conditions the prevalence has substantially increased following large scale migration from Africa and the Middle East to Europe. The hemoglobin diseases severely limit both, life expectancy and quality of life and require either life-long supportive therapy if cure cannot be achieved by allogeneic stem cell transplantation. Strategies for ex vivo gene therapy aiming at either re-establishing normal ß-globin chain synthesis or at re-activating fetal γ-globin chain and HbF expression are currently in clinical development. The European Medicine Agency (EMA) conditionally licensed gene addition therapy based on lentiviral transduction of hematopoietic stem cells in 2019 for a selected group of patients with transfusion dependent non-ß° thalassemia major without a suitable stem cell donor. Gene therapy thus offers a relevant chance to this group of patients for whom cure has previously not been on the horizon. In this review, we discuss the potential and the challenges of gene addition and gene editing strategies for the hemoglobin diseases.
Copyright © 2020 the Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the European Hematology Association.
Figures
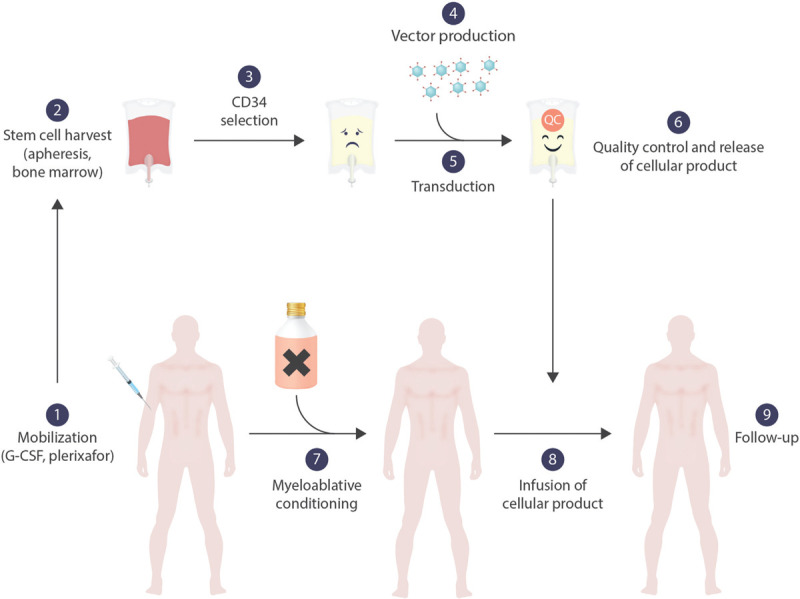
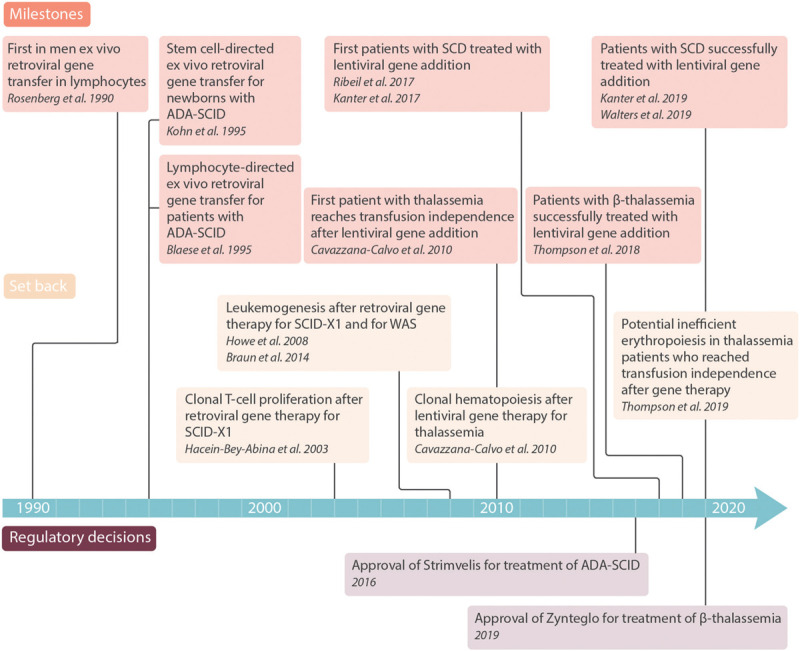
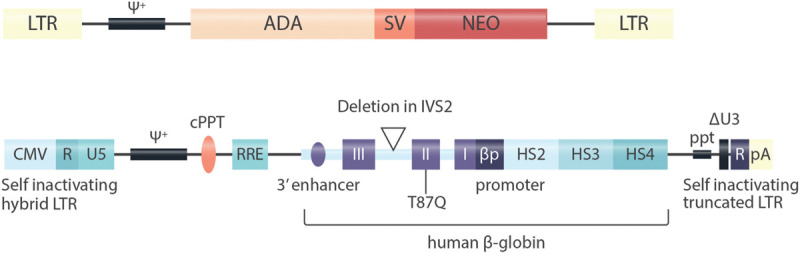
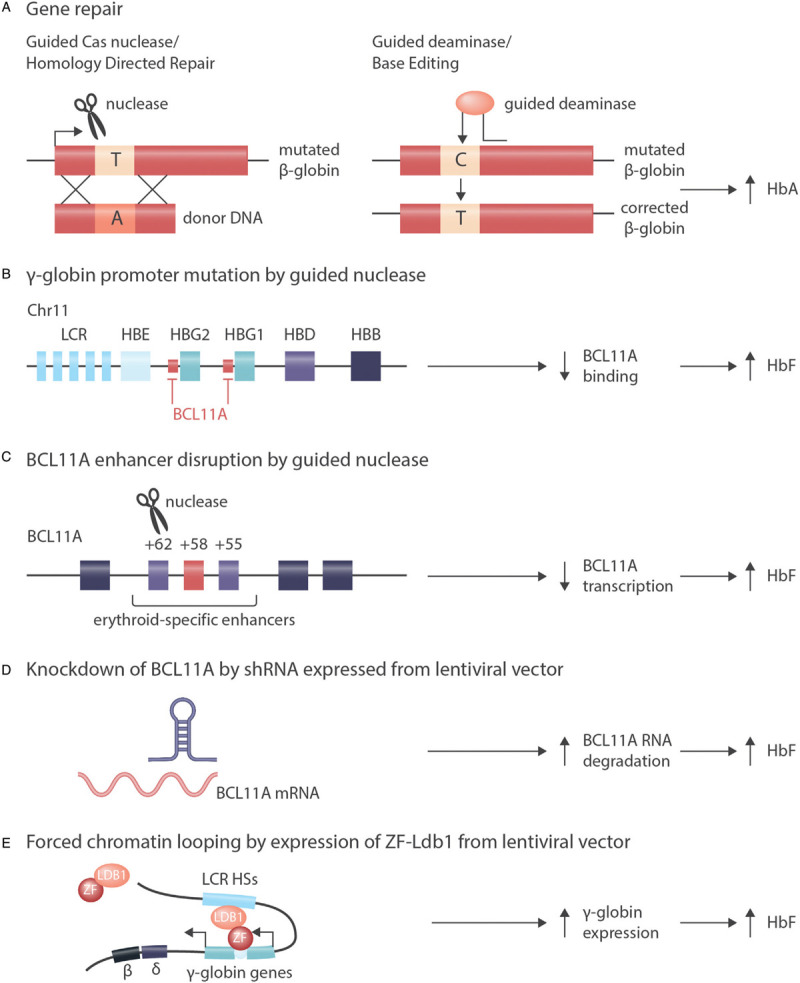
Similar articles
-
Hemoglobin disorders: lentiviral gene therapy in the starting blocks to enter clinical practice.Exp Hematol. 2018 Aug;64:12-32. doi: 10.1016/j.exphem.2018.05.004. Epub 2018 May 26. Exp Hematol. 2018. PMID: 29807062 Review.
-
Gene therapy of hemoglobinopathies: progress and future challenges.Hum Mol Genet. 2019 Oct 1;28(R1):R24-R30. doi: 10.1093/hmg/ddz172. Hum Mol Genet. 2019. PMID: 31322165 Review.
-
Disorders of the synthesis of human fetal hemoglobin.IUBMB Life. 2008 Feb;60(2):94-111. doi: 10.1002/iub.4. IUBMB Life. 2008. PMID: 18379999 Review.
-
Gene therapy in thalassemia and hemoglobinopathies.Mediterr J Hematol Infect Dis. 2009 Nov 13;1(1):e2009008. doi: 10.4084/MJHID.2009.008. Mediterr J Hematol Infect Dis. 2009. PMID: 21415990 Free PMC article.
-
Cell and Gene Therapy for the Beta-Thalassemias: Advances and Prospects.Hum Gene Ther. 2016 Apr;27(4):295-304. doi: 10.1089/hum.2016.037. Hum Gene Ther. 2016. PMID: 27021486 Free PMC article. Review.
Cited by
-
Viral and Non-Viral Systems to Deliver Gene Therapeutics to Clinical Targets.Int J Mol Sci. 2024 Jul 4;25(13):7333. doi: 10.3390/ijms25137333. Int J Mol Sci. 2024. PMID: 39000440 Free PMC article. Review.
-
Sickle Cell Disease.Transfus Med Hemother. 2024 Aug 6;51(5):332-344. doi: 10.1159/000540149. eCollection 2024 Oct. Transfus Med Hemother. 2024. PMID: 39371249 Free PMC article. Review.
-
Gene Editing-Based Technologies for Beta-hemoglobinopathies Treatment.Biology (Basel). 2022 Jun 4;11(6):862. doi: 10.3390/biology11060862. Biology (Basel). 2022. PMID: 35741383 Free PMC article. Review.
-
Does the world need germline editing for β-thalassemia?Haematologica. 2022 Jun 1;107(6):1235-1236. doi: 10.3324/haematol.2021.279998. Haematologica. 2022. PMID: 34706499 Free PMC article. No abstract available.
-
Selecting β-thalassemia Patients for Gene Therapy: A Decision-making Algorithm.Hemasphere. 2021 Apr 29;5(5):e555. doi: 10.1097/HS9.0000000000000555. eCollection 2021 May. Hemasphere. 2021. PMID: 33969274 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials