

Interrogating Genomic-Scale Data to Resolve Recalcitrant Nodes in the Spider Tree of Life
- PMID: 32986823
- PMCID: PMC7947752
- DOI: 10.1093/molbev/msaa251
Interrogating Genomic-Scale Data to Resolve Recalcitrant Nodes in the Spider Tree of Life
Abstract
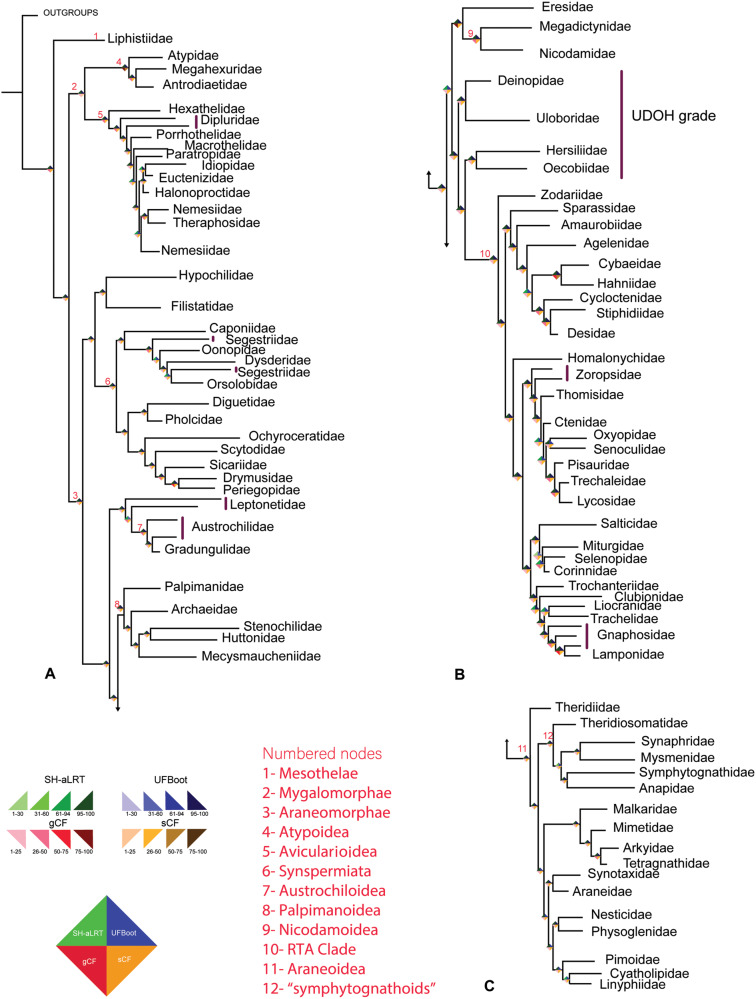
Genome-scale data sets are converging on robust, stable phylogenetic hypotheses for many lineages; however, some nodes have shown disagreement across classes of data. We use spiders (Araneae) as a system to identify the causes of incongruence in phylogenetic signal between three classes of data: exons (as in phylotranscriptomics), noncoding regions (included in ultraconserved elements [UCE] analyses), and a combination of both (as in UCE analyses). Gene orthologs, coded as amino acids and nucleotides (with and without third codon positions), were generated by querying published transcriptomes for UCEs, recovering 1,931 UCE loci (codingUCEs). We expected that congeners represented in the codingUCE and UCEs data would form clades in the presence of phylogenetic signal. Noncoding regions derived from UCE sequences were recovered to test the stability of relationships. Phylogenetic relationships resulting from all analyses were largely congruent. All nucleotide data sets from transcriptomes, UCEs, or a combination of both recovered similar topologies in contrast with results from transcriptomes analyzed as amino acids. Most relationships inferred from low-occupancy data sets, containing several hundreds of loci, were congruent across Araneae, as opposed to high occupancy data matrices with fewer loci, which showed more variation. Furthermore, we found that low-occupancy data sets analyzed as nucleotides (as is typical of UCE data sets) can result in more congruent relationships than high occupancy data sets analyzed as amino acids (as in phylotranscriptomics). Thus, omitting data, through amino acid translation or via retention of only high occupancy loci, may have a deleterious effect in phylogenetic reconstruction.
Keywords: Araneae; noncoding regions; phylogeny; target-capture; transcriptomics.
© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures




References
-
- Arnedo MA, Hormiga G, Scharff N.. 2009. Higher-level phylogenetics of linyphiid spiders (Araneae, Linyphiidae) based on morphological and molecular evidence. Cladistics 25(3):231–262. - PubMed
-
- Babb PL, Lahens NF, Correa-Garhwal SM, Nicholson DN, Kim EJ, Hogenesch JB, Kuntner M, Higgins L, Hayashi CY, Agnarsson I, et al.2017. The Nephila clavipes genome highlights the diversity of spider silk genes and their complex expression. Nat Genet. 49(6):895–903. - PubMed
-
- Ballesteros JA, Sharma P.2019. A critical appraisal of the placement of Xiphosura (Chelicerata) with account of known sources of phylogenetic error. Syst Biol. 68(6):896–917. - PubMed
-
- Bond JE, Garrison NL, Hamilton CA, Godwin RL, Hedin M, Agnarsson I.. 2014. Phylogenomics resolves a spider backbone phylogeny and rejects a prevailing paradigm for orb web evolution. Curr Biol. 24(15):1765–1771. - PubMed
-
- Bossert S, Murray EA, Almeida EAB, Brady SG, Blaimer BB, Danforth BN.. 2019. Combining transcriptomes and ultraconserved elements to illuminate the phylogeny of Apidae. Mol Phylogenet Evol. 130:121–131. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
