

Control of craniofacial and brain development by Cullin3-RING ubiquitin ligases: Lessons from human disease genetics
- PMID: 32986984
- PMCID: PMC10627151
- DOI: 10.1016/j.yexcr.2020.112300
Control of craniofacial and brain development by Cullin3-RING ubiquitin ligases: Lessons from human disease genetics
Abstract
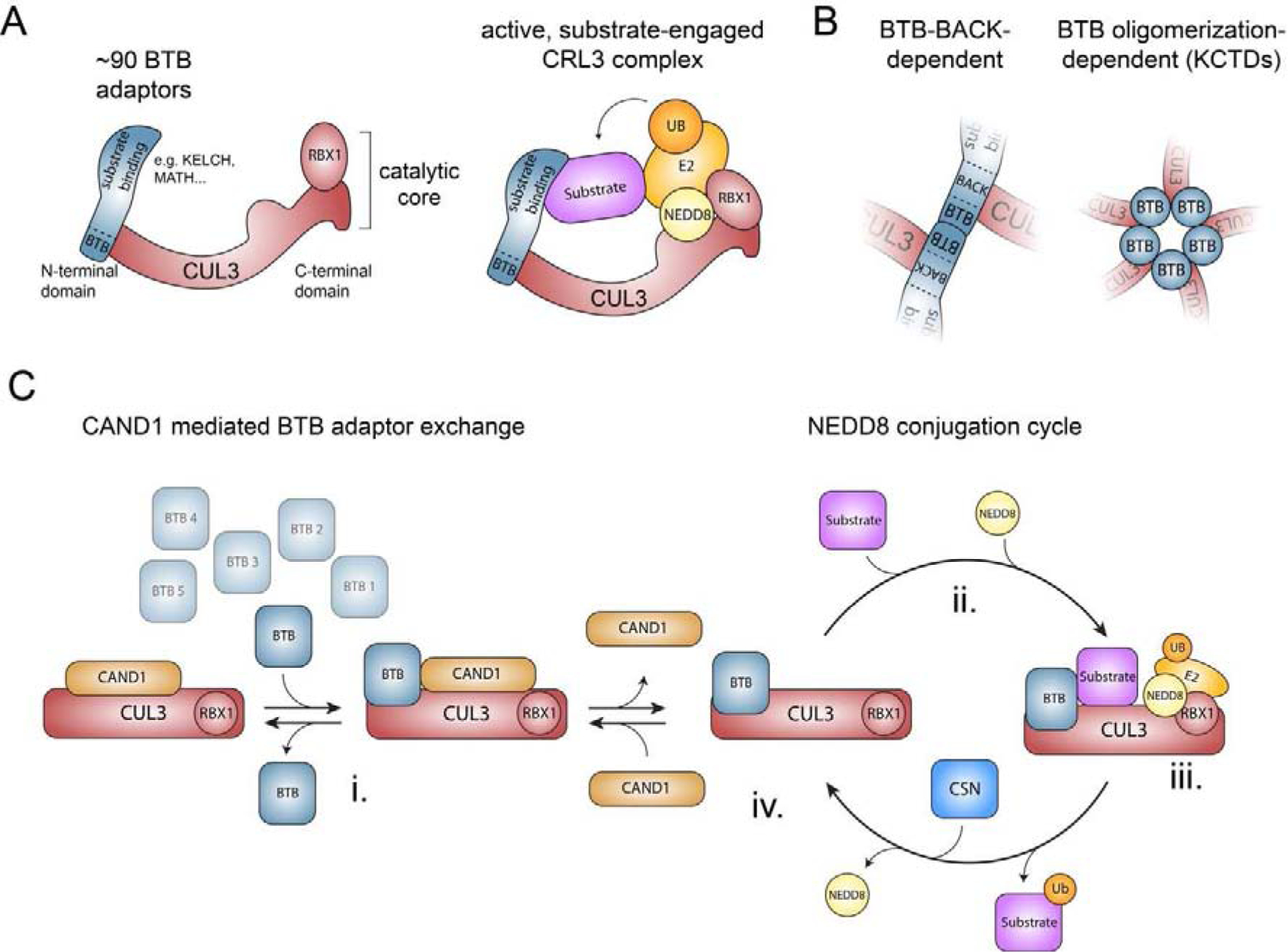
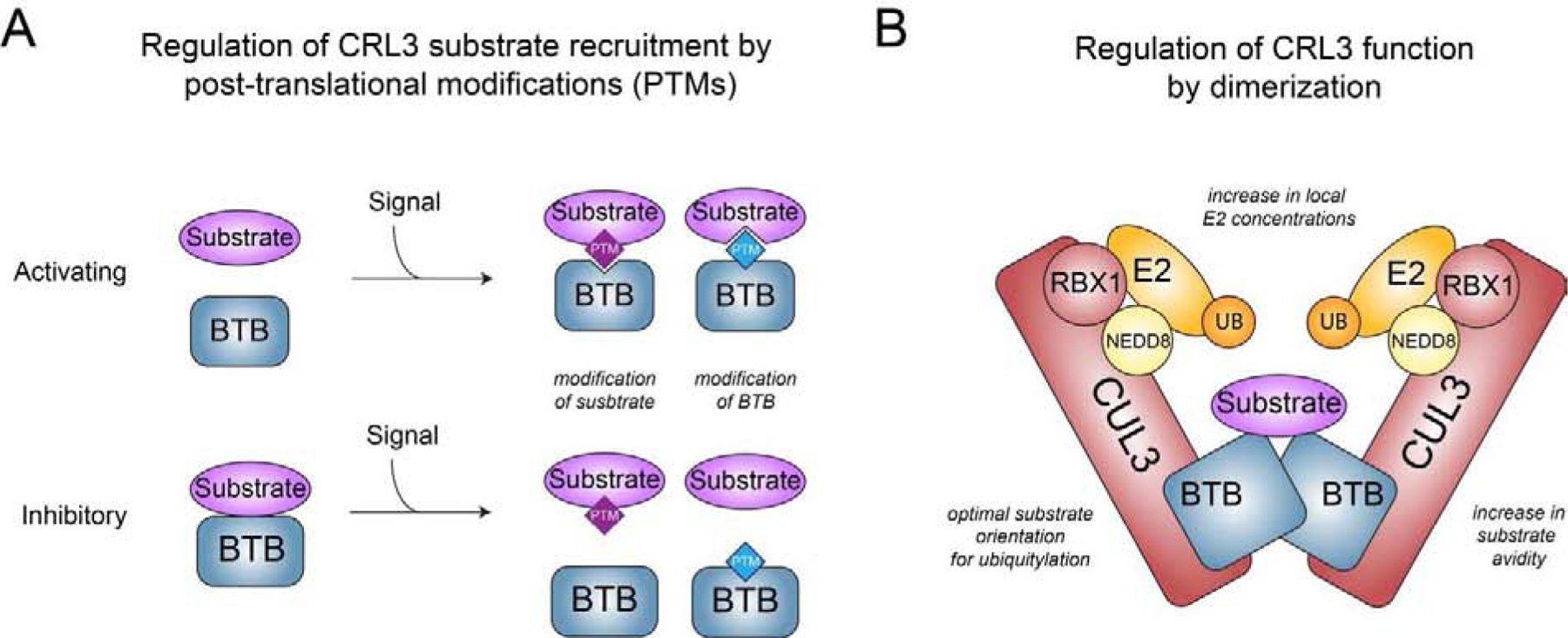
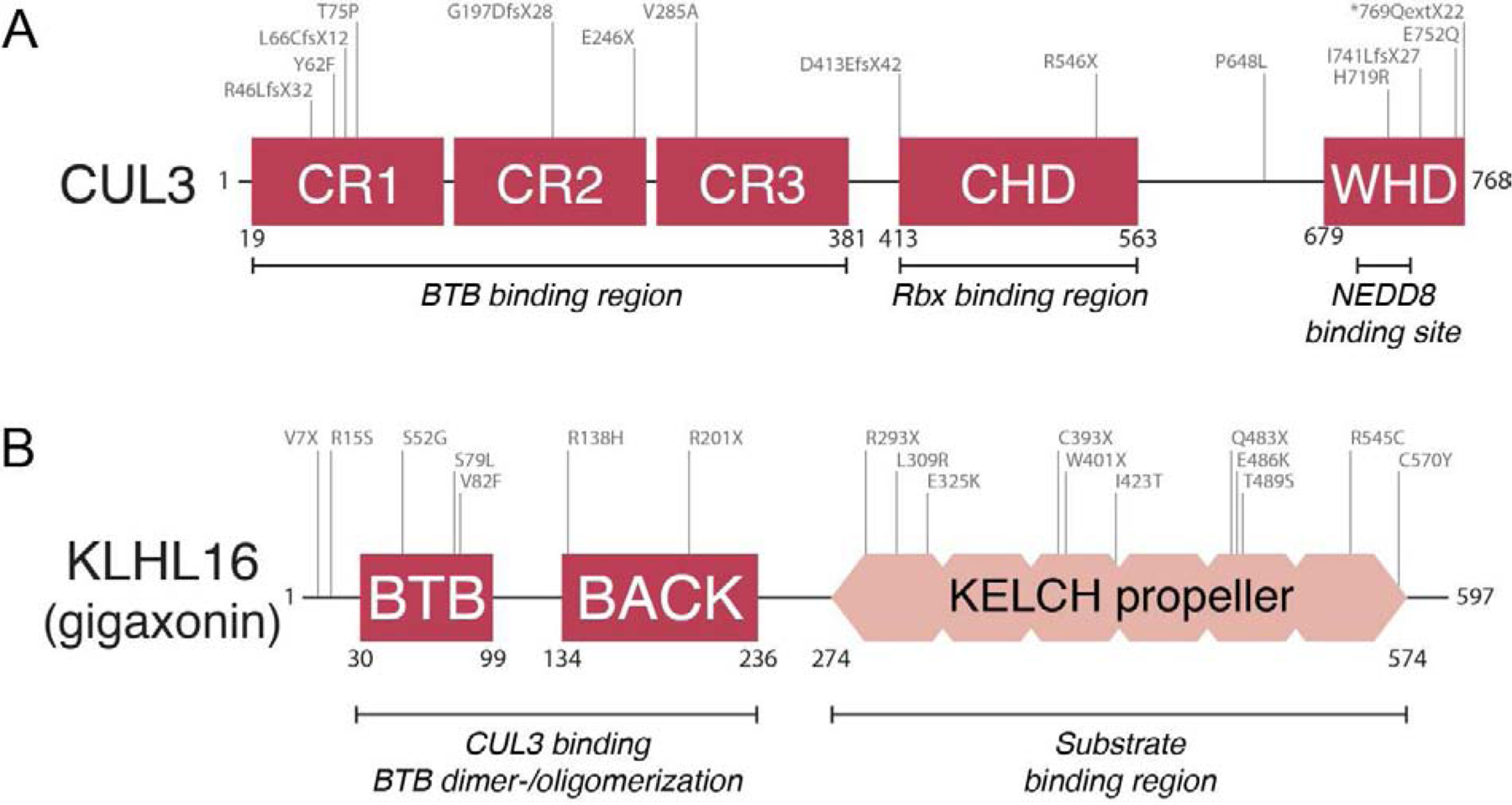
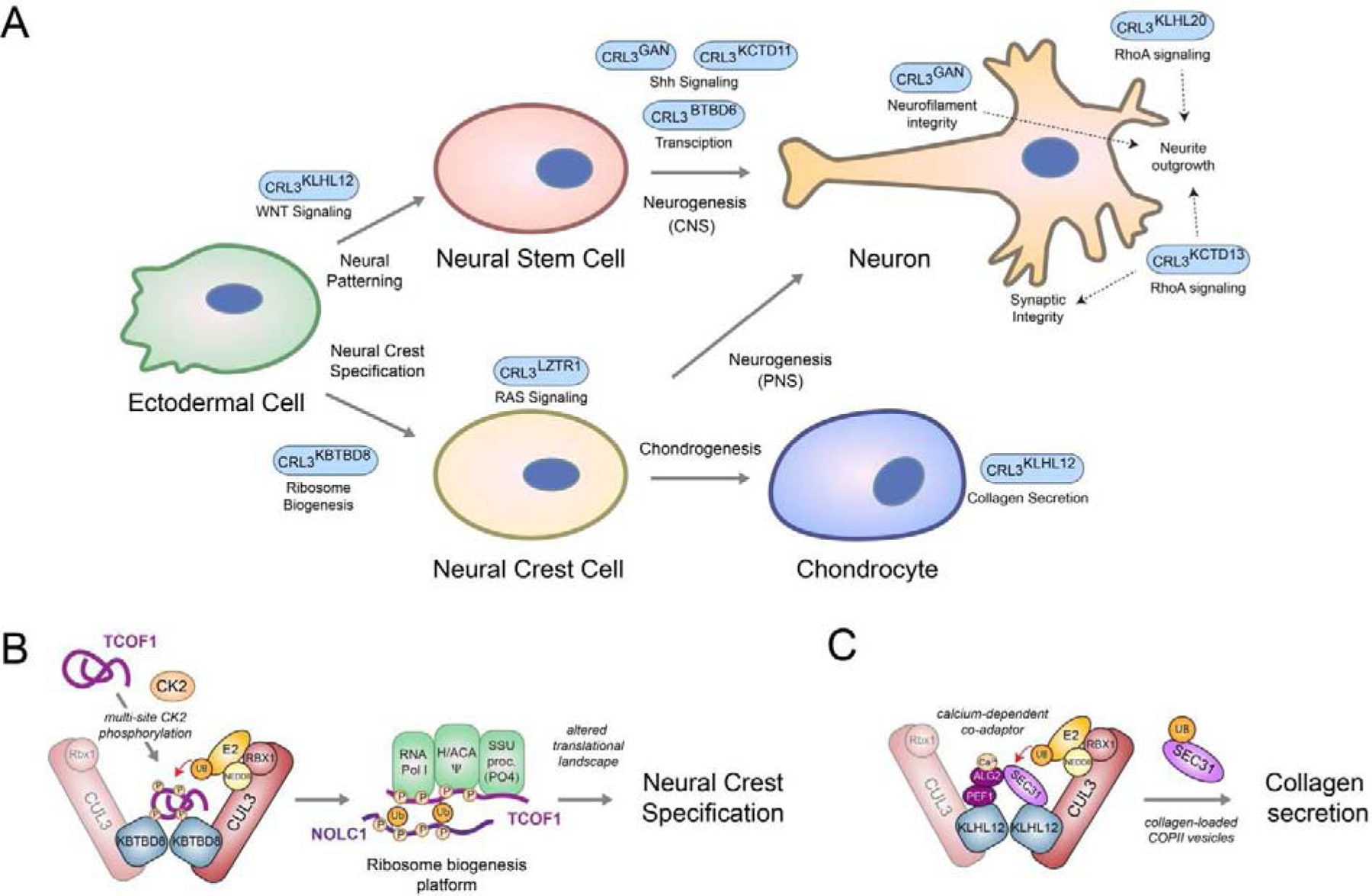
Metazoan development relies on intricate cell differentiation, communication, and migration pathways, which ensure proper formation of specialized cell types, tissues, and organs. These pathways are crucially controlled by ubiquitylation, a reversible post-translational modification that regulates the stability, activity, localization, or interaction landscape of substrate proteins. Specificity of ubiquitylation is ensured by E3 ligases, which bind substrates and co-operate with E1 and E2 enzymes to mediate ubiquitin transfer. Cullin3-RING ligases (CRL3s) are a large class of multi-subunit E3s that have emerged as important regulators of cell differentiation and development. In particular, recent evidence from human disease genetics, animal models, and mechanistic studies have established their involvement in the control of craniofacial and brain development. Here, we summarize regulatory principles of CRL3 assembly, substrate recruitment, and ubiquitylation that allow this class of E3s to fulfill their manifold functions in development. We further review our current mechanistic understanding of how specific CRL3 complexes orchestrate neuroectodermal differentiation and highlight diseases associated with their dysregulation. Based on evidence from human disease genetics, we propose that other unknown CRL3 complexes must help coordinate craniofacial and brain development and discuss how combining emerging strategies from the field of disease gene discovery with biochemical and human pluripotent stem cell approaches will likely facilitate their identification.
Keywords: BTB; Brain development; CRL; CRL3; Craniofacial development; Developmental diseases; Ubiquitin.
Published by Elsevier Inc.
Conflict of interest statement
Conflict of Interest:
The authors declare no conflict of interest.
Figures
References
-
- Clague MJ, Urbe S, Komander D, Breaking the chains: deubiquitylating enzyme specificity begets function, Nat Rev Mol Cell Biol 20 (2019) 338–352. - PubMed
-
- Oh E, Akopian D, Rape M, Principles of Ubiquitin-Dependent Signaling, Annu Rev Cell Dev Biol 34 (2018) 137–162. - PubMed
-
- Komander D, Rape M, The ubiquitin code, Annu Rev Biochem 81 (2012) 203–229. - PubMed
-
- Yau R, Rape M, The increasing complexity of the ubiquitin code, Nat Cell Biol 18 (2016) 579–586. - PubMed
-
- Di Fiore PP, Polo S, Hofmann K, When ubiquitin meets ubiquitin receptors: a signalling connection, Nat Rev Mol Cell Biol 4 (2003) 491–497. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
