

An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer's disease
- PMID: 32989324
- PMCID: PMC8098004
- DOI: 10.1038/s41588-020-0696-0
An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer's disease
Erratum in
-
Author Correction: An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer's disease.Nat Genet. 2020 Nov;52(11):1266. doi: 10.1038/s41588-020-00733-7. Nat Genet. 2020. PMID: 33033384 Free PMC article.
Abstract
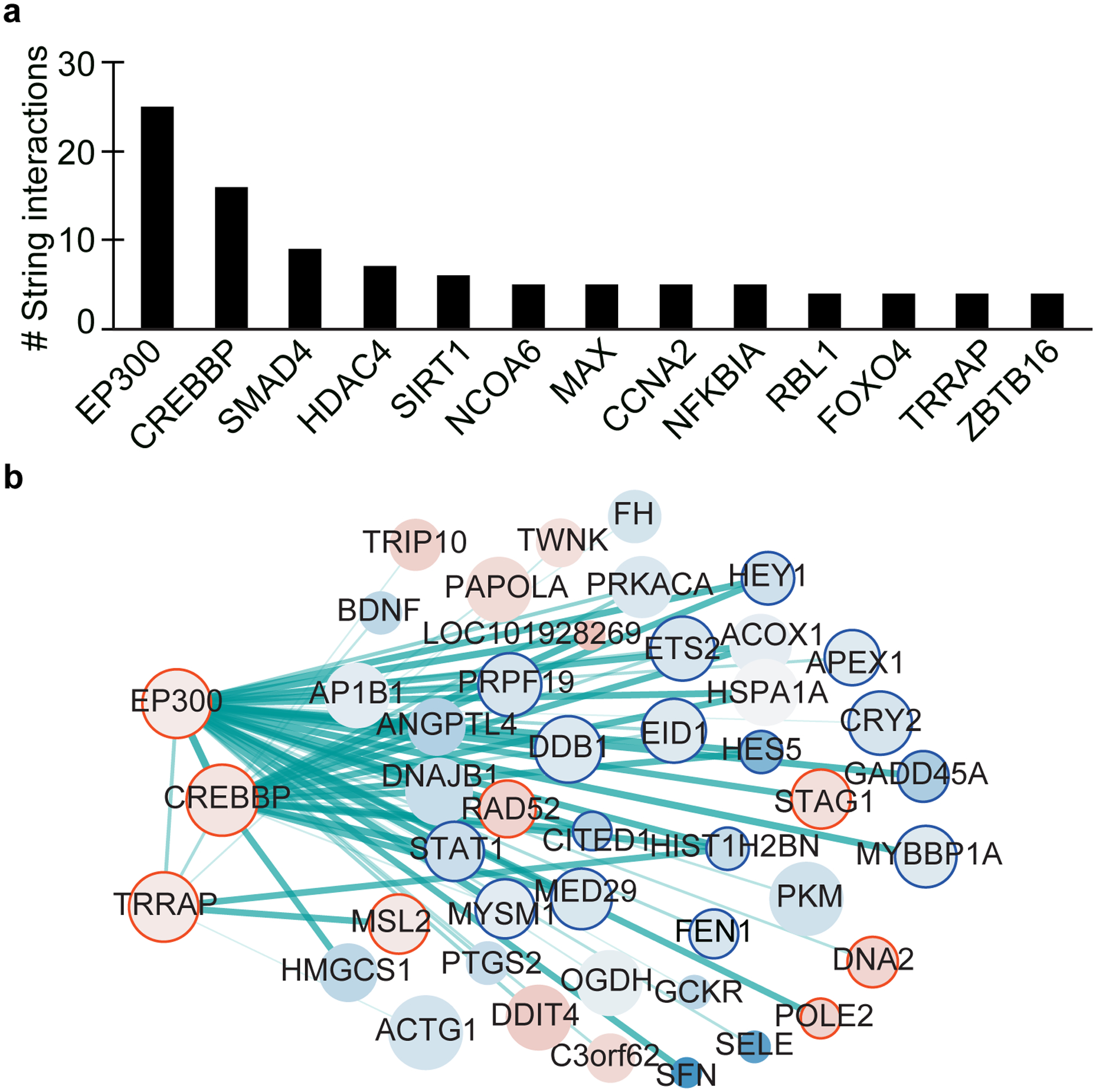
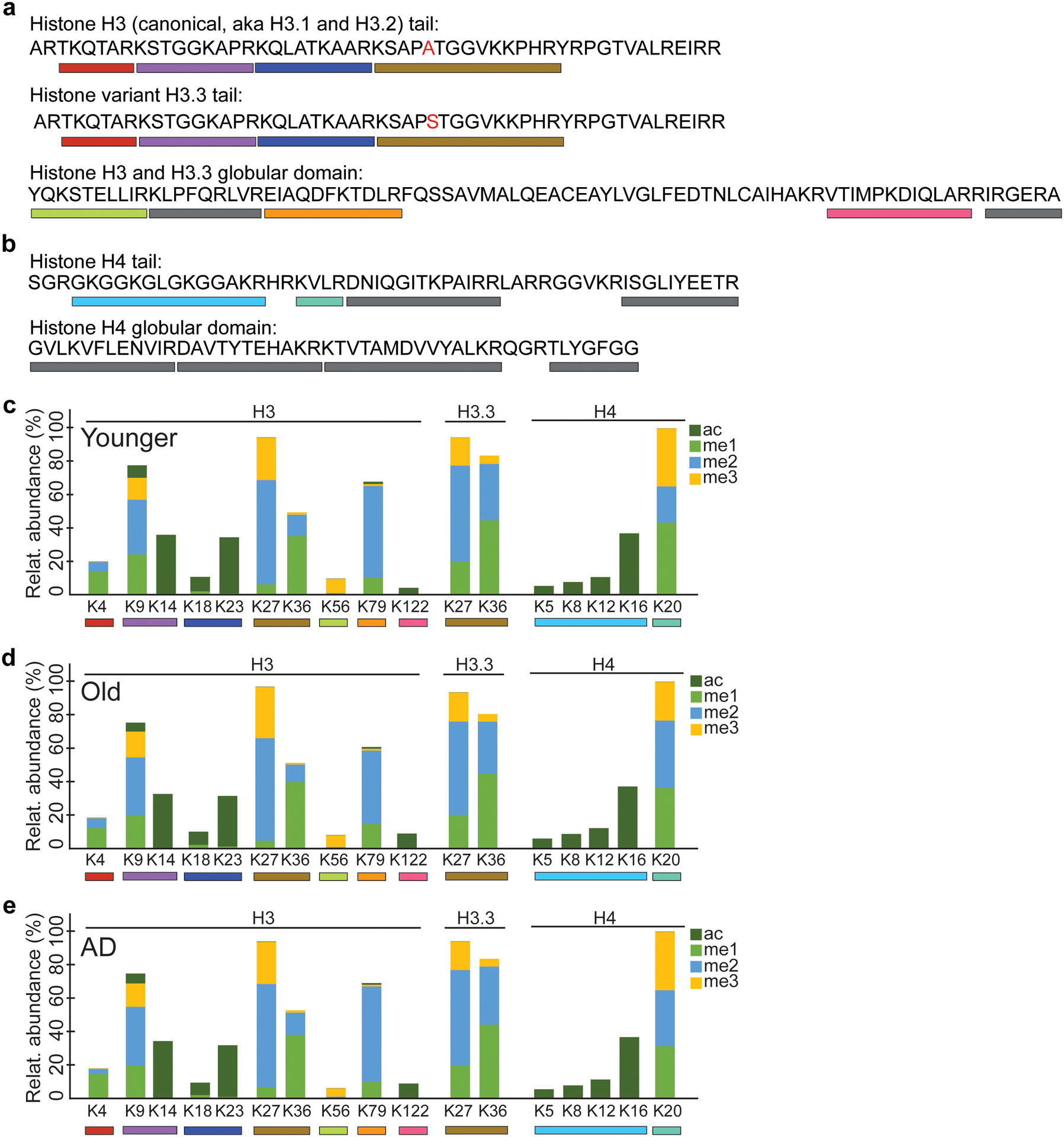
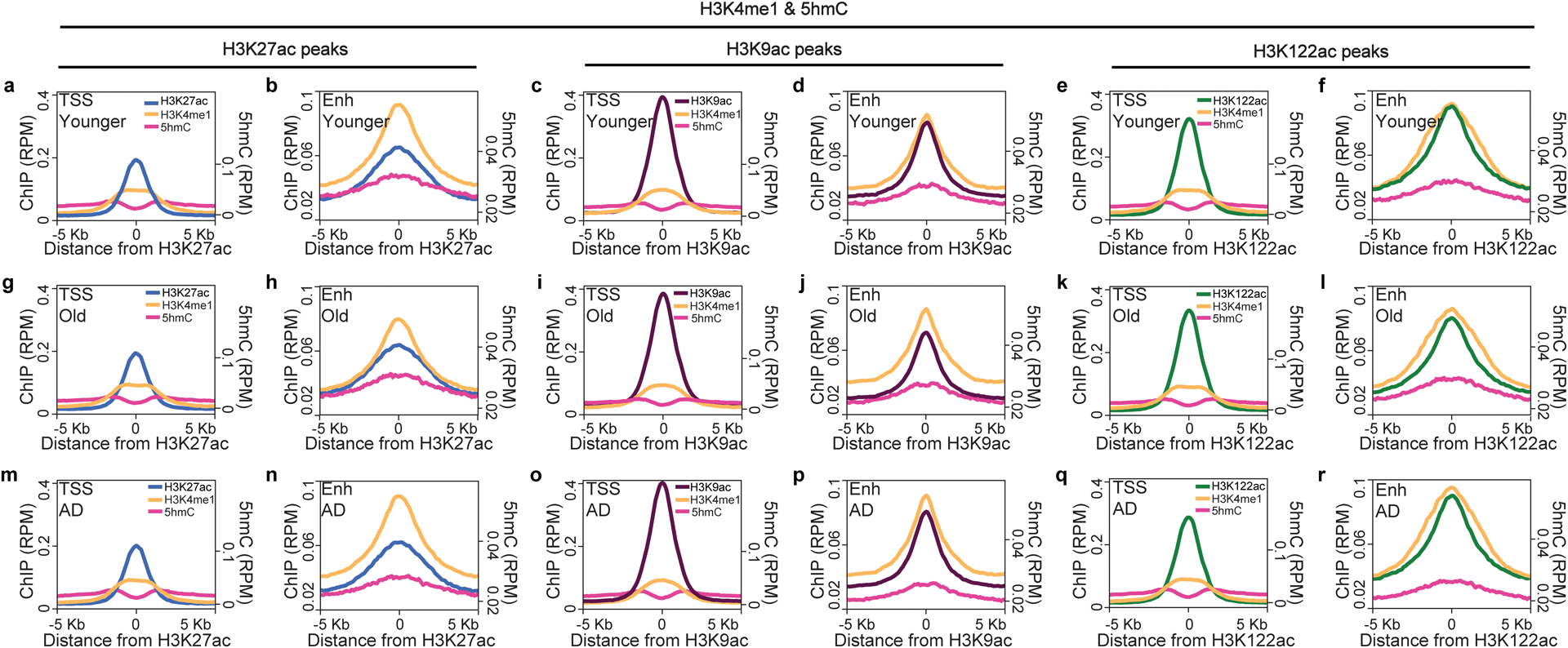
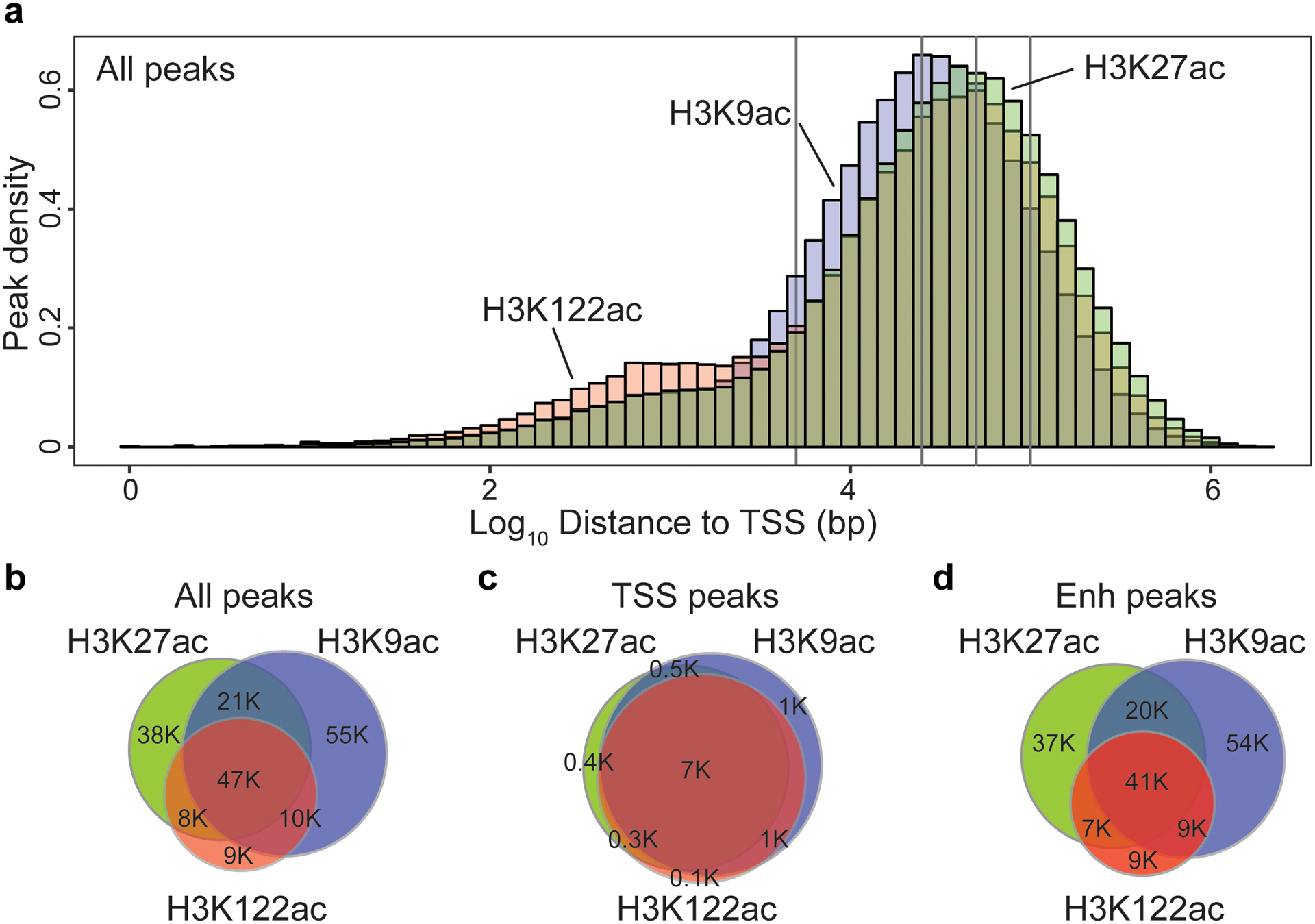
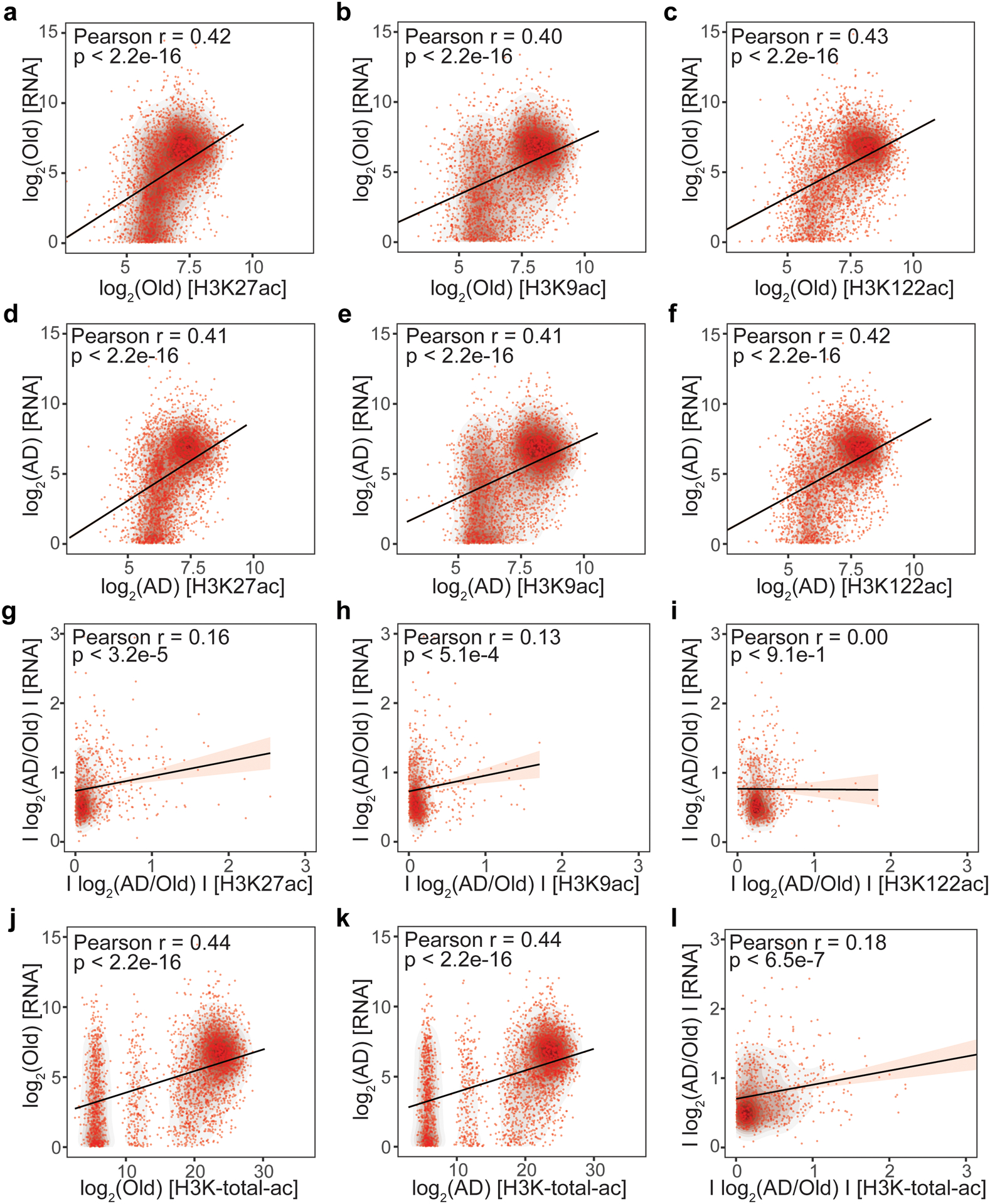
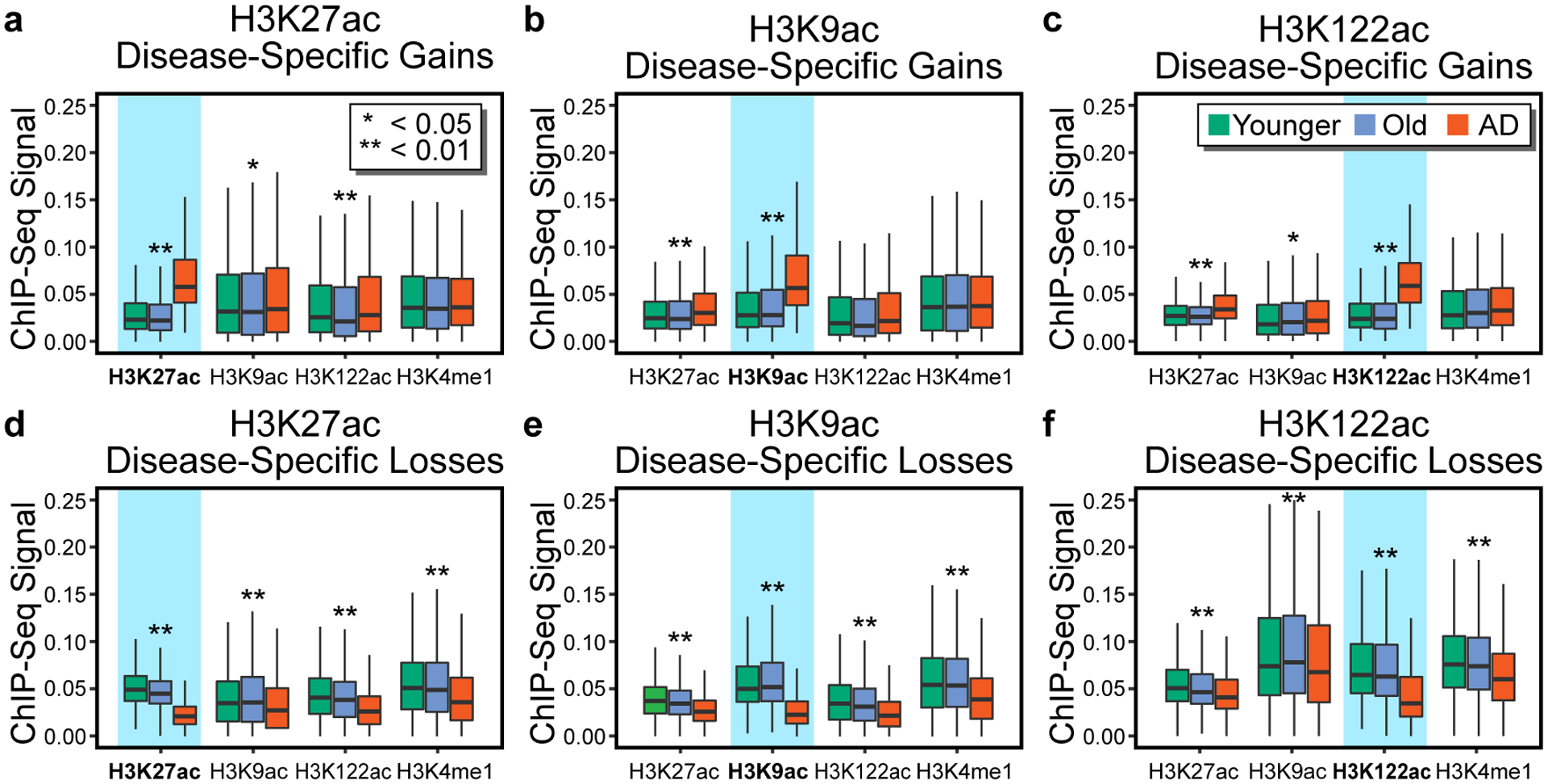
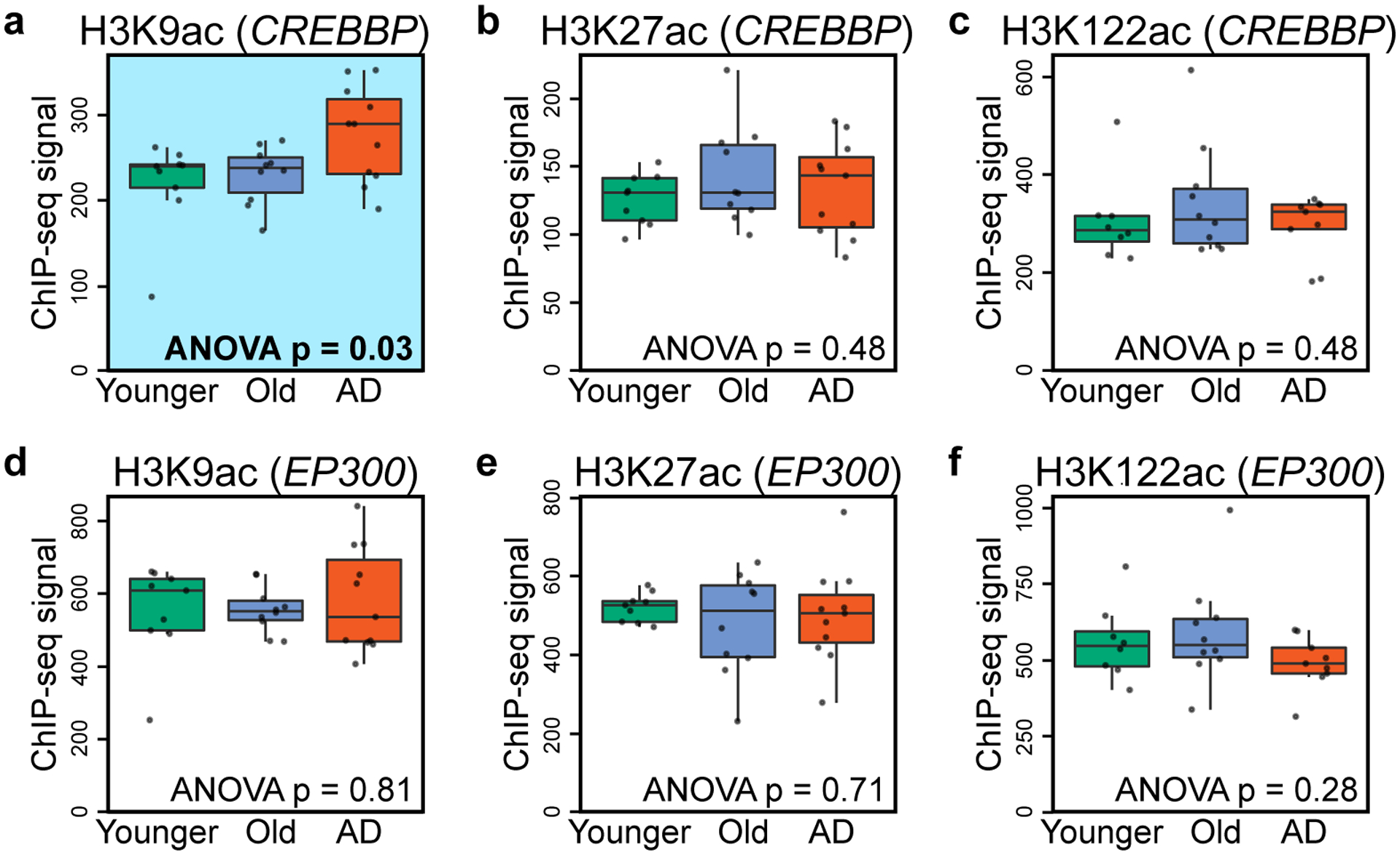
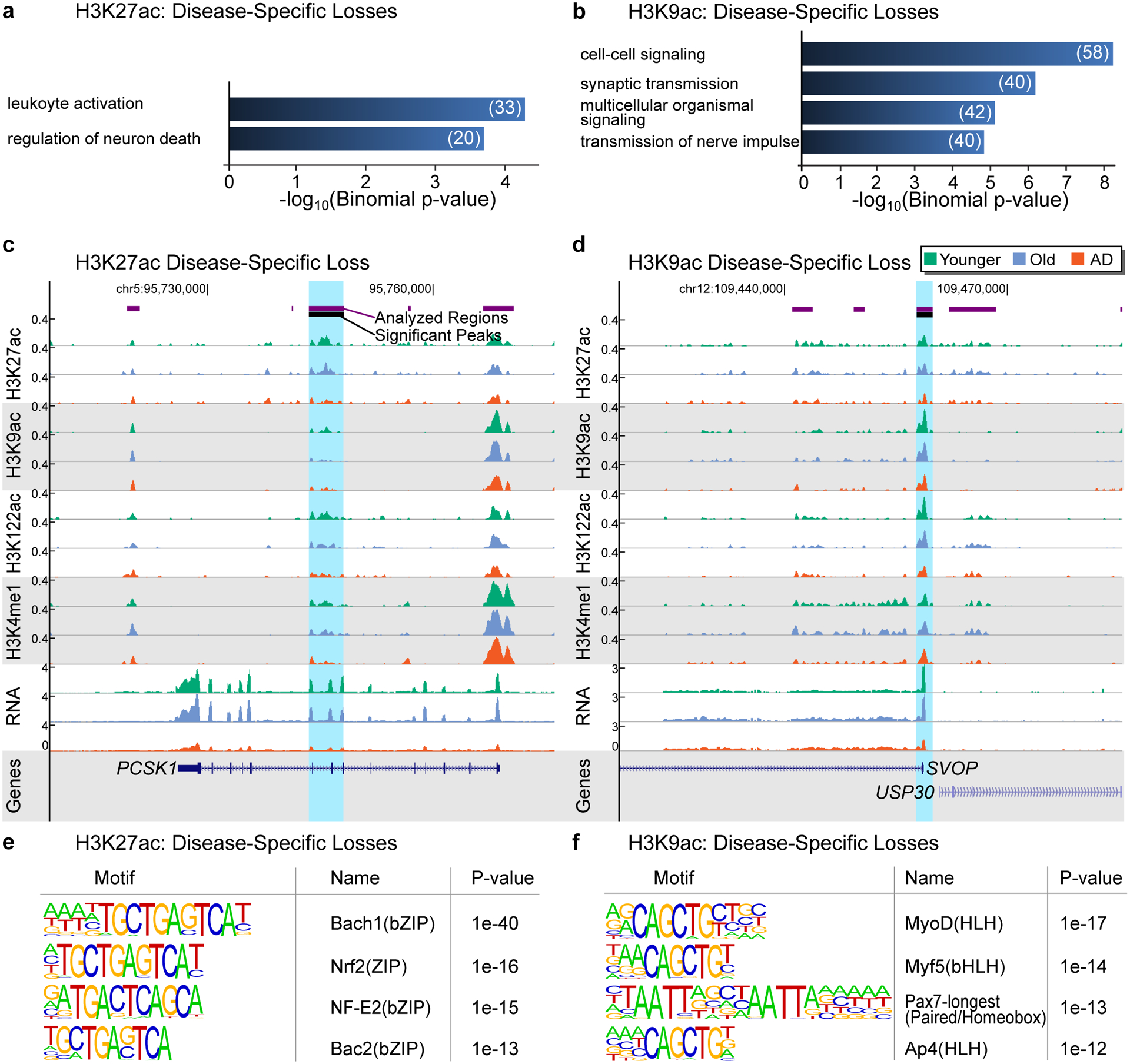
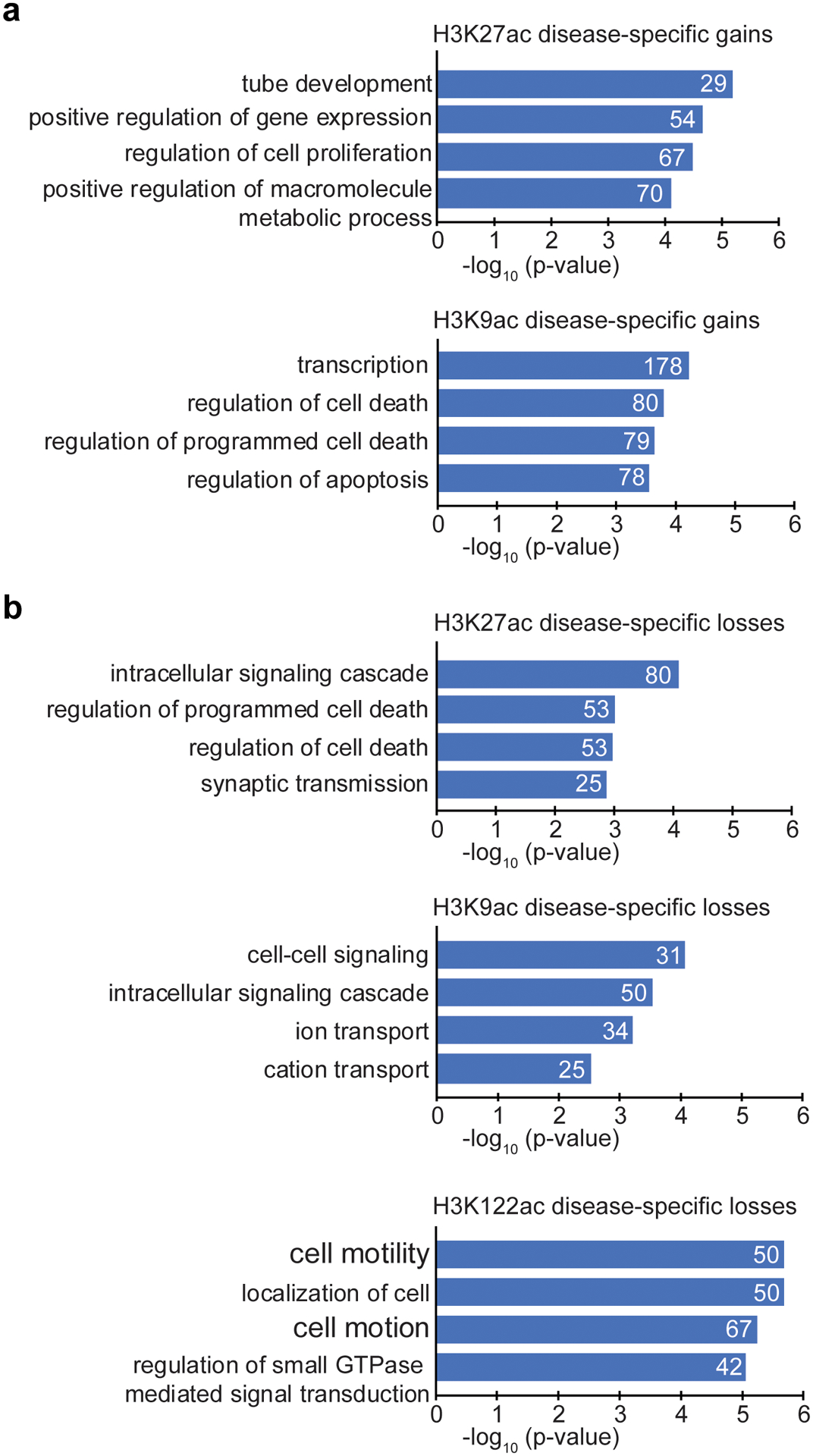
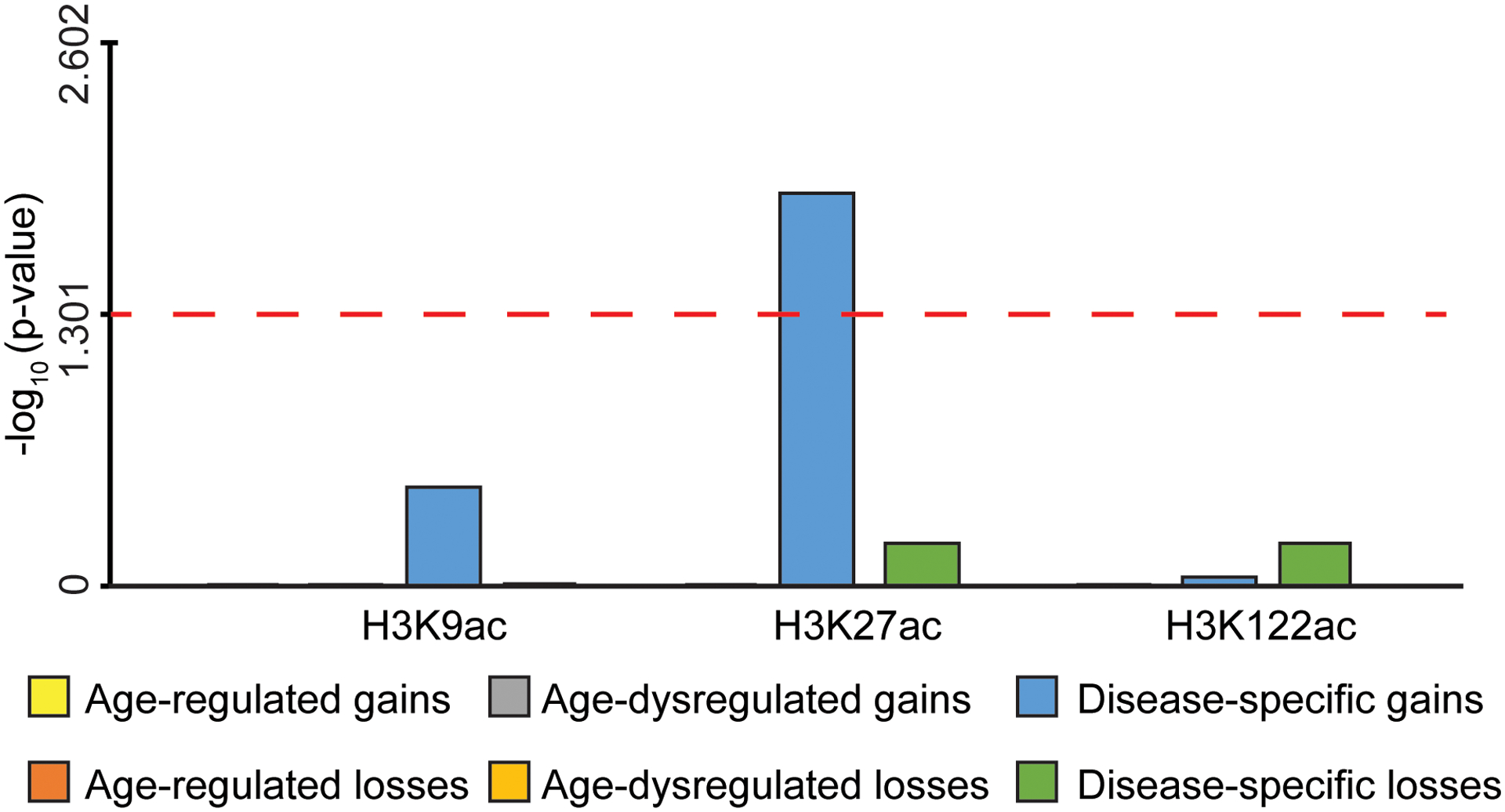
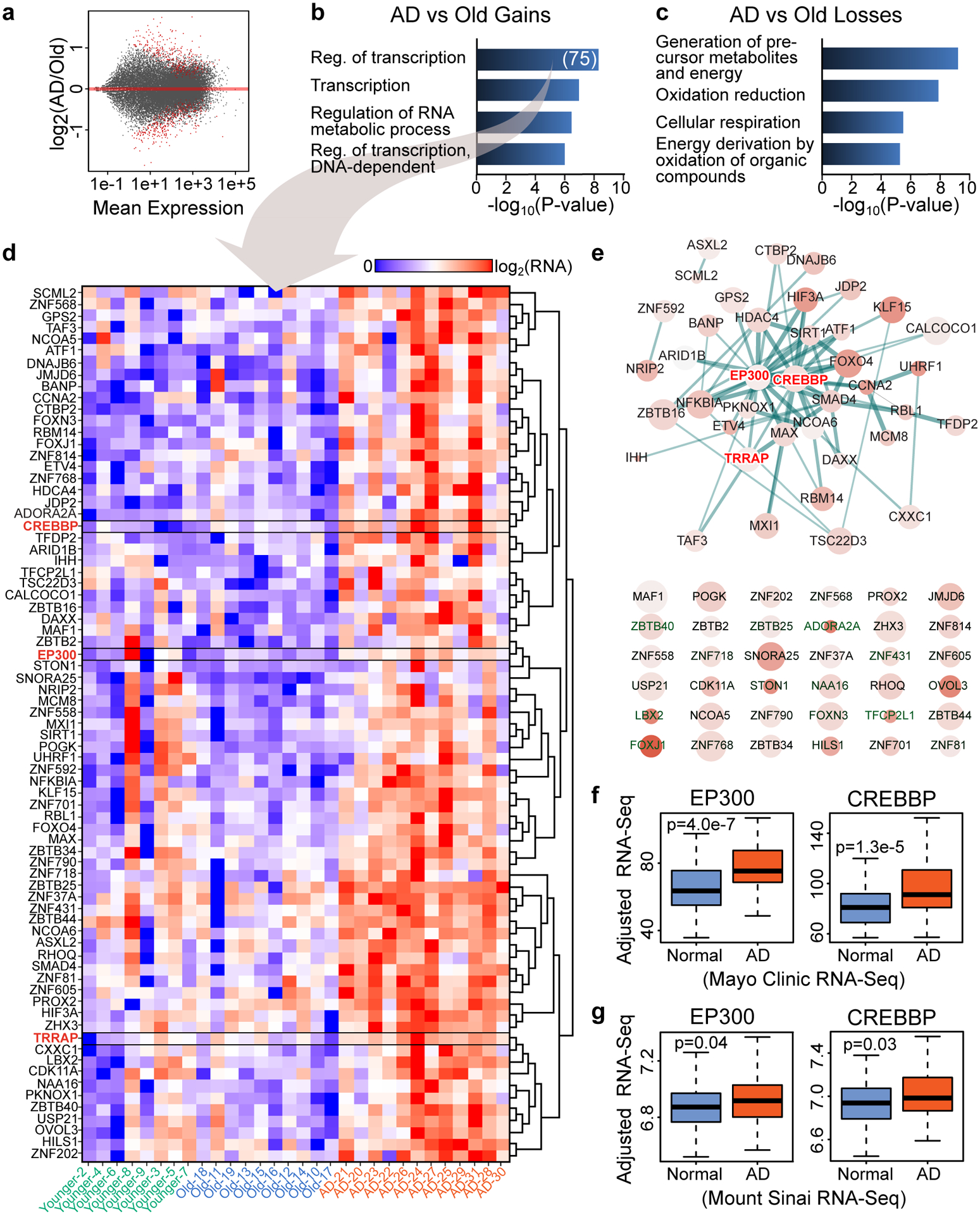
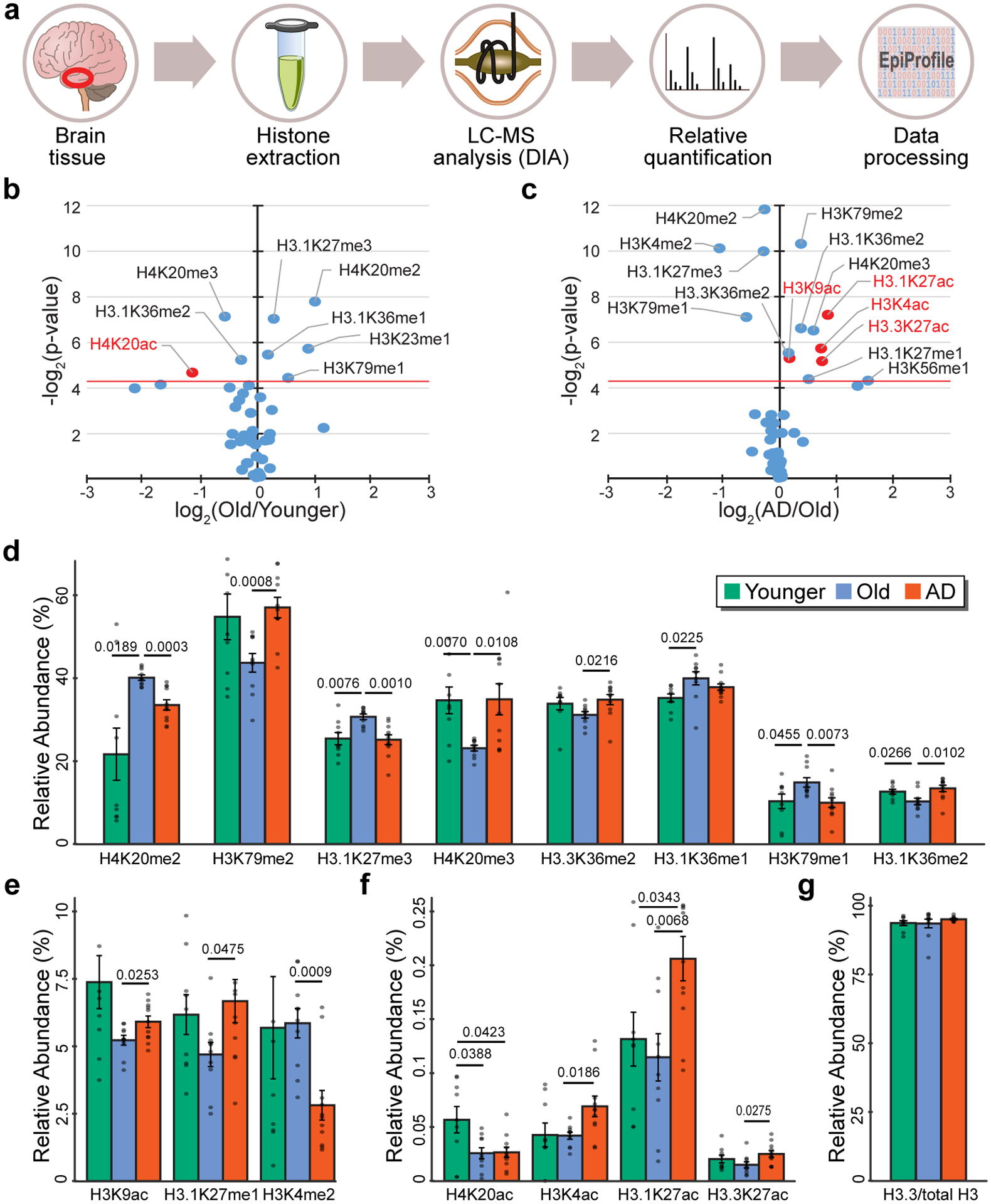
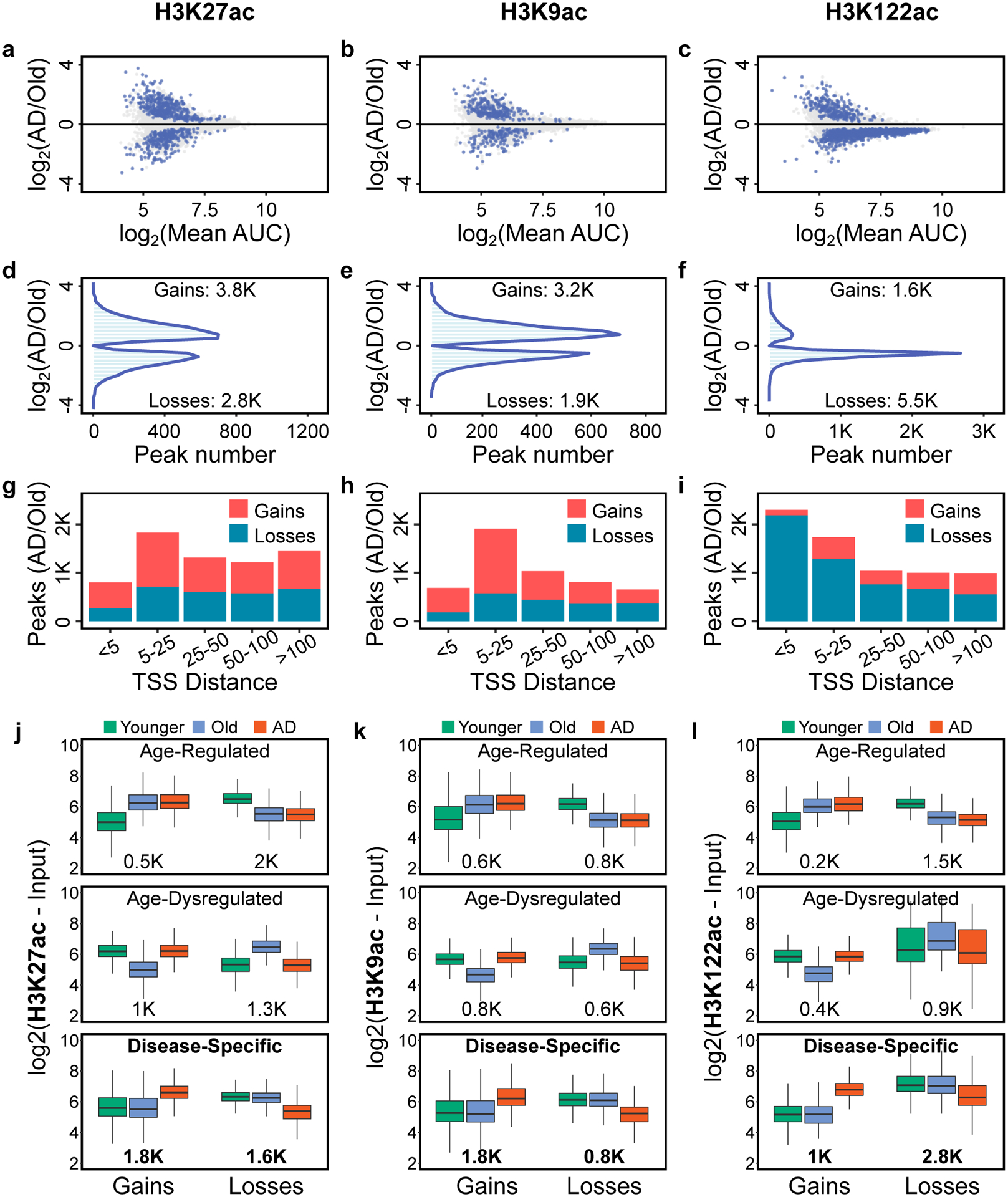
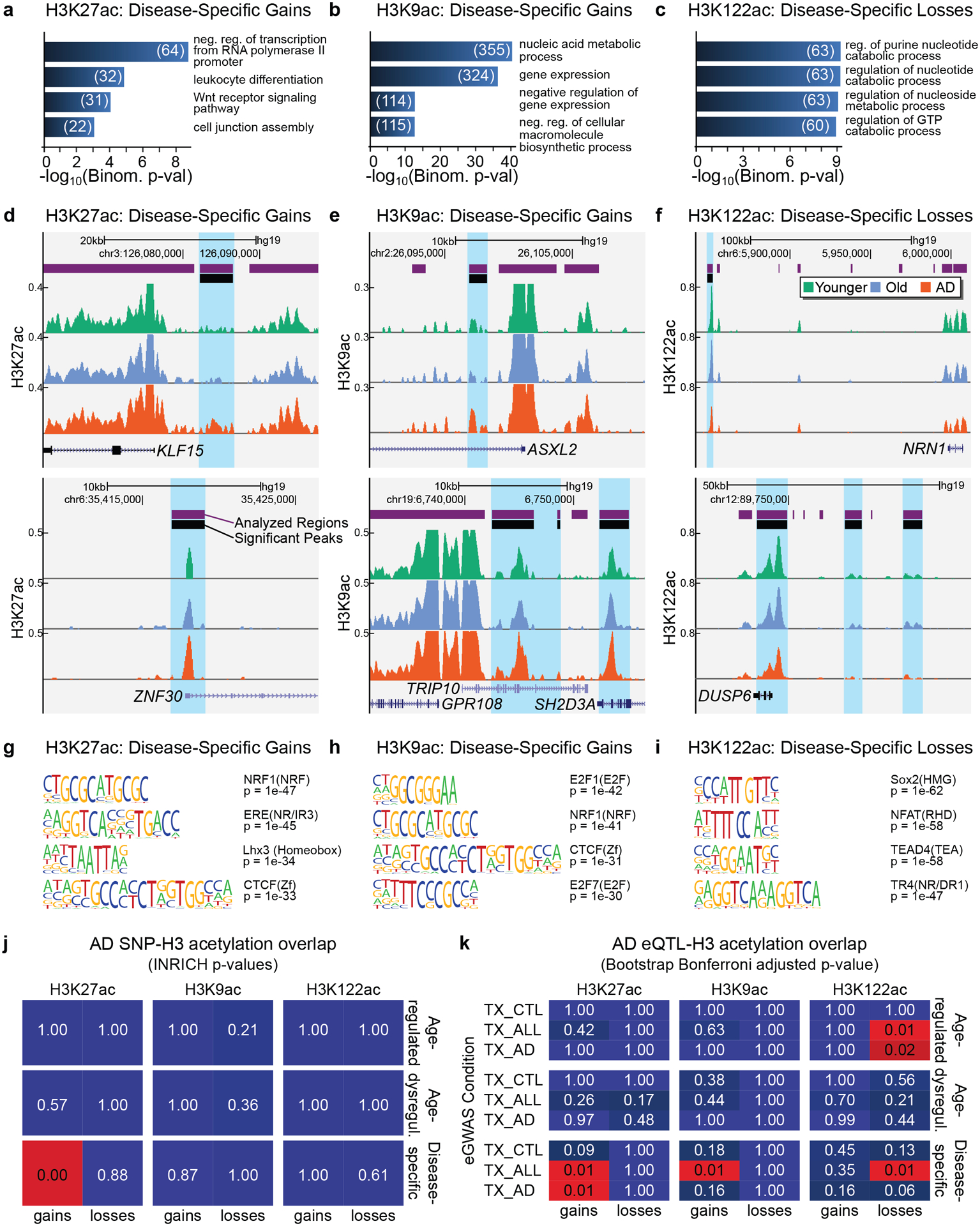
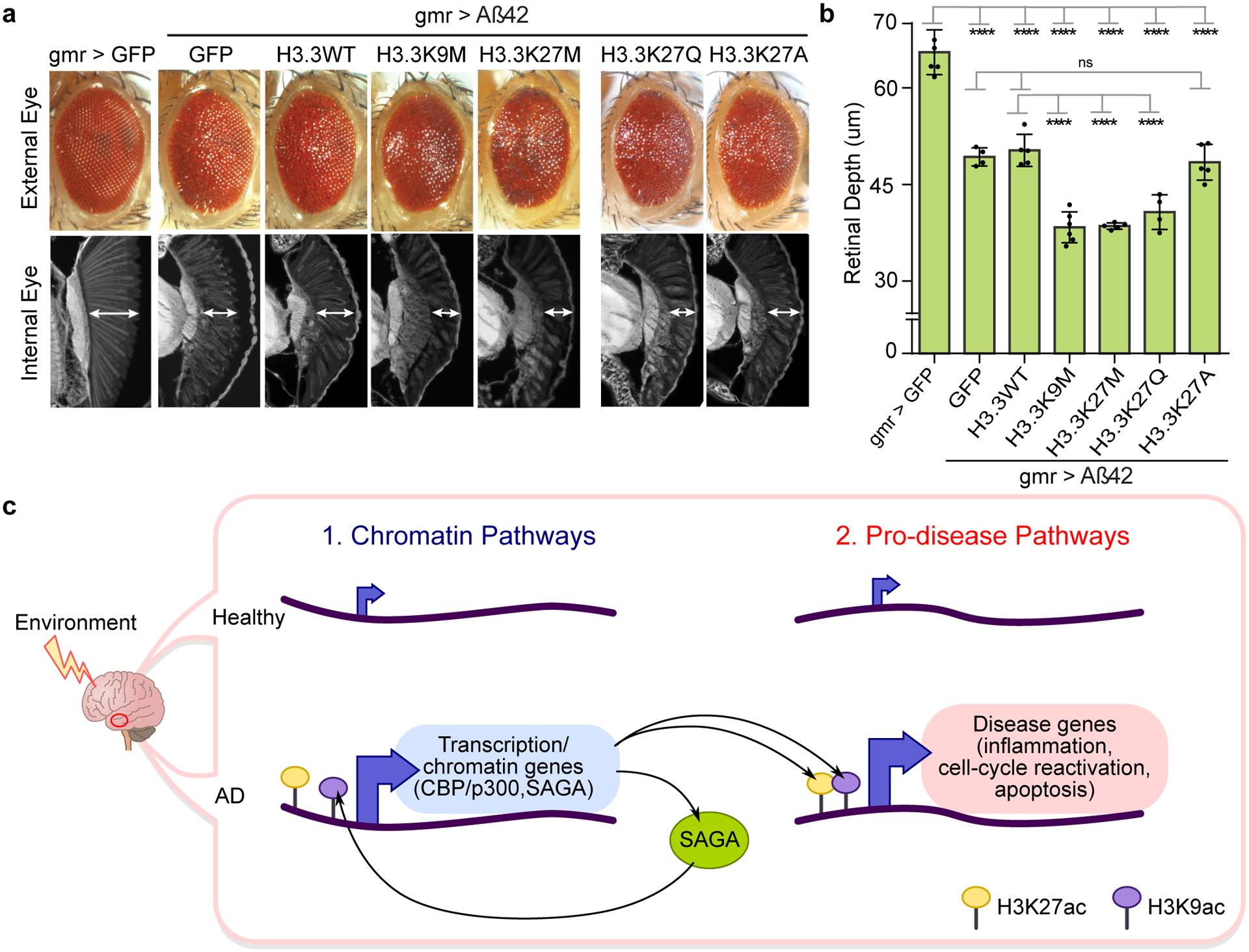
Protein aggregation is the hallmark of neurodegeneration, but the molecular mechanisms underlying late-onset Alzheimer's disease (AD) are unclear. Here we integrated transcriptomic, proteomic and epigenomic analyses of postmortem human brains to identify molecular pathways involved in AD. RNA sequencing analysis revealed upregulation of transcription- and chromatin-related genes, including the histone acetyltransferases for H3K27ac and H3K9ac. An unbiased proteomic screening singled out H3K27ac and H3K9ac as the main enrichments specific to AD. In turn, epigenomic profiling revealed gains in the histone H3 modifications H3K27ac and H3K9ac linked to transcription, chromatin and disease pathways in AD. Increasing genome-wide H3K27ac and H3K9ac in a fly model of AD exacerbated amyloid-β42-driven neurodegeneration. Together, these findings suggest that AD involves a reconfiguration of the epigenome, wherein H3K27ac and H3K9ac affect disease pathways by dysregulating transcription- and chromatin-gene feedback loops. The identification of this process highlights potential epigenetic strategies for early-stage disease treatment.
Conflict of interest statement
Competing interests
C.H. holds a patent on technology used (US8741567) and is a shareholder in Shanghai Epican Genetech LTD.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
