

Mutations disrupting neuritogenesis genes confer risk for cerebral palsy
- PMID: 32989326
- PMCID: PMC9148538
- DOI: 10.1038/s41588-020-0695-1
Mutations disrupting neuritogenesis genes confer risk for cerebral palsy
Erratum in
-
Author Correction: Mutations disrupting neuritogenesis genes confer risk for cerebral palsy.Nat Genet. 2021 Mar;53(3):412. doi: 10.1038/s41588-021-00780-8. Nat Genet. 2021. PMID: 33432185 No abstract available.
Abstract
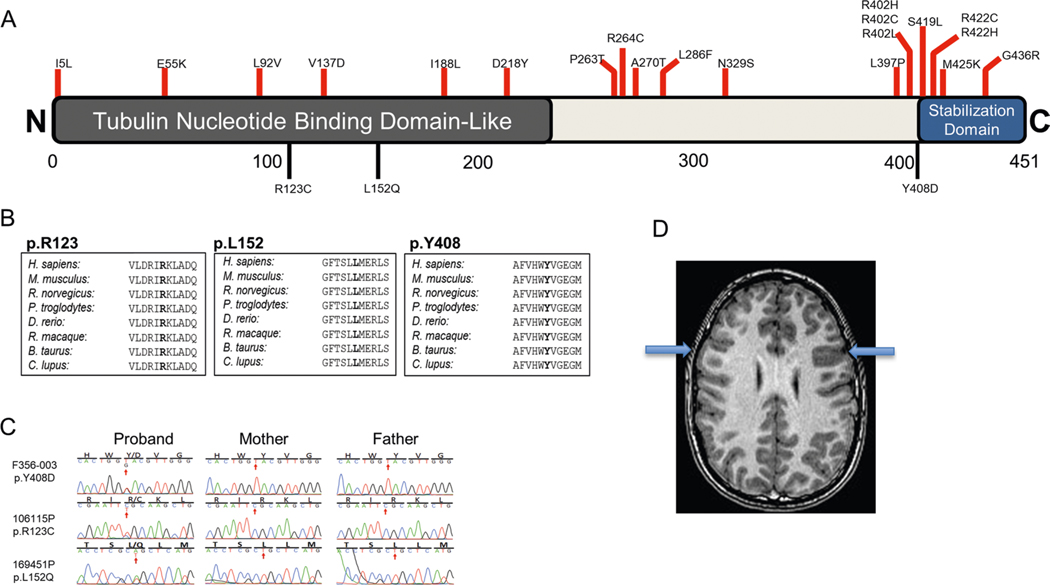
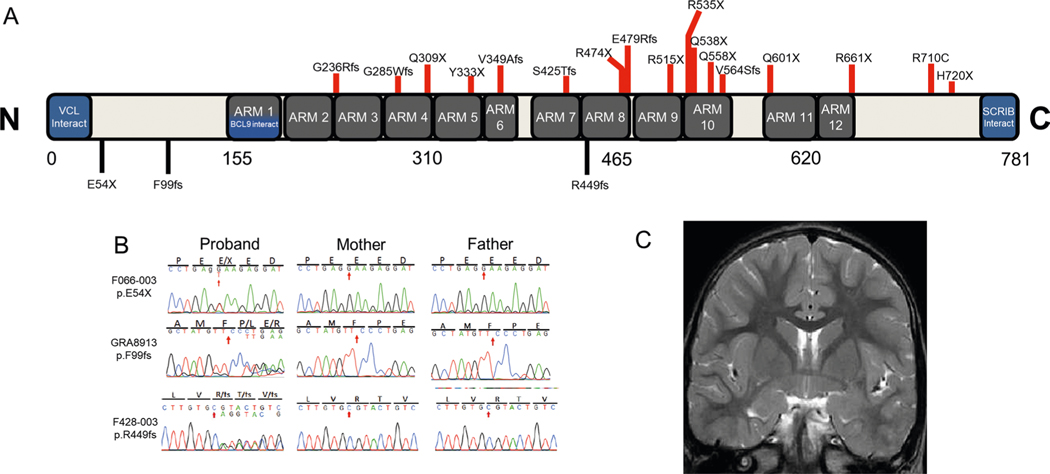
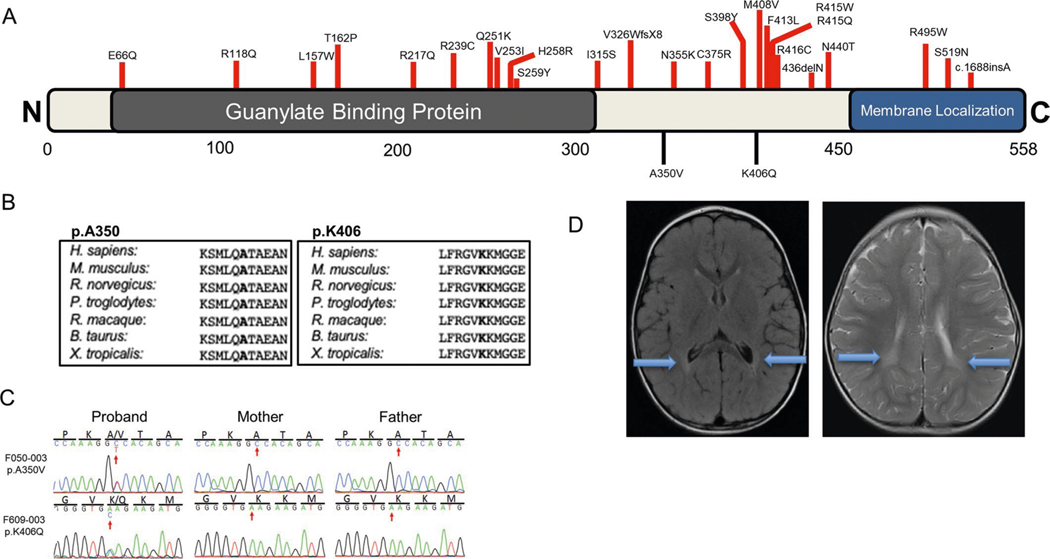
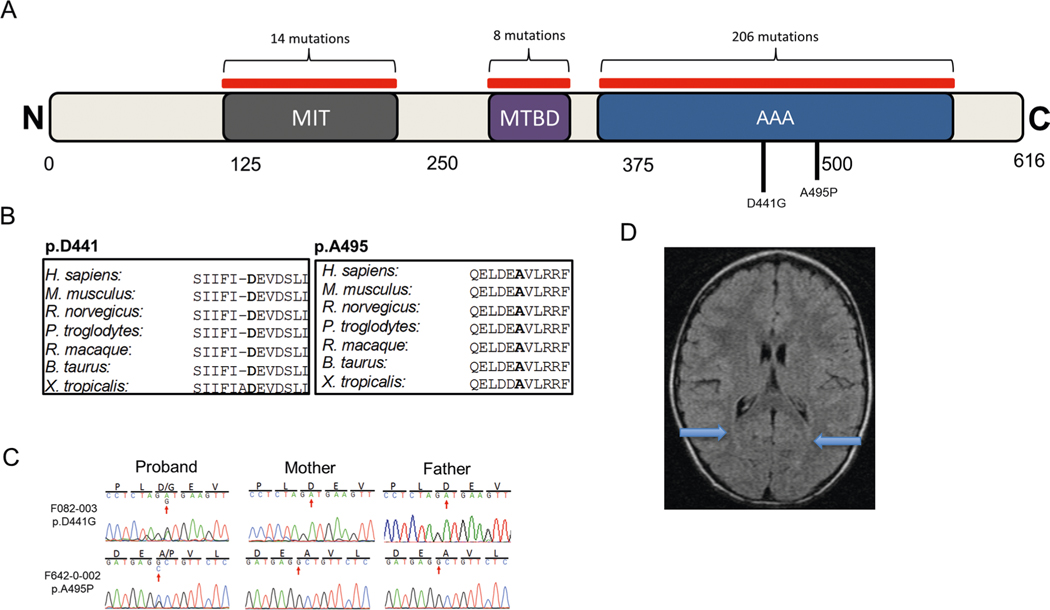
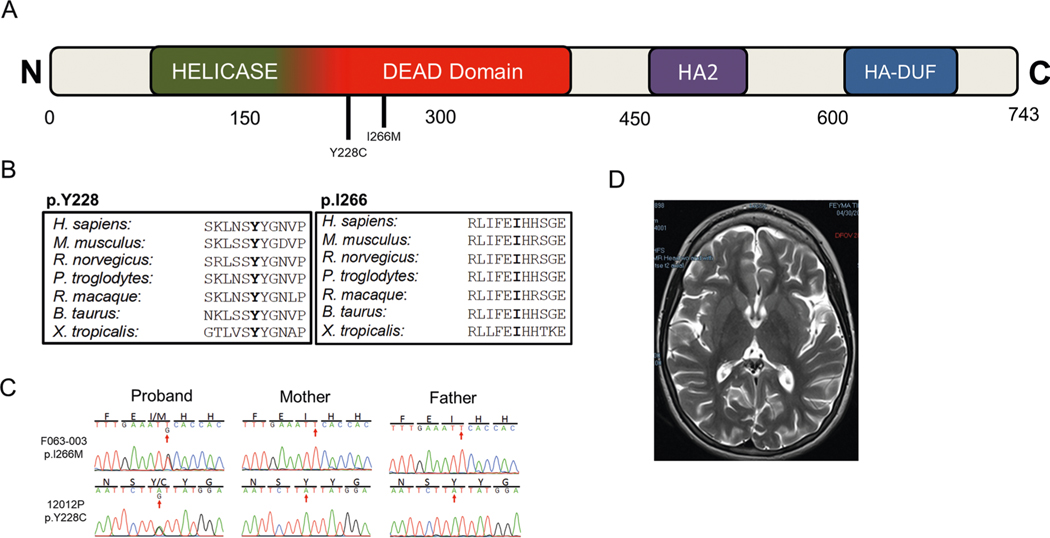
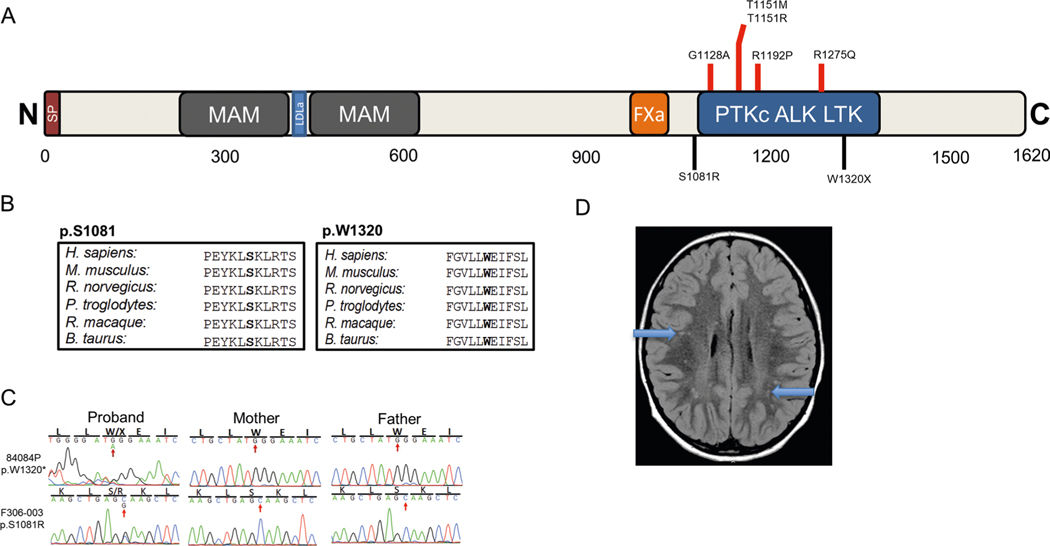
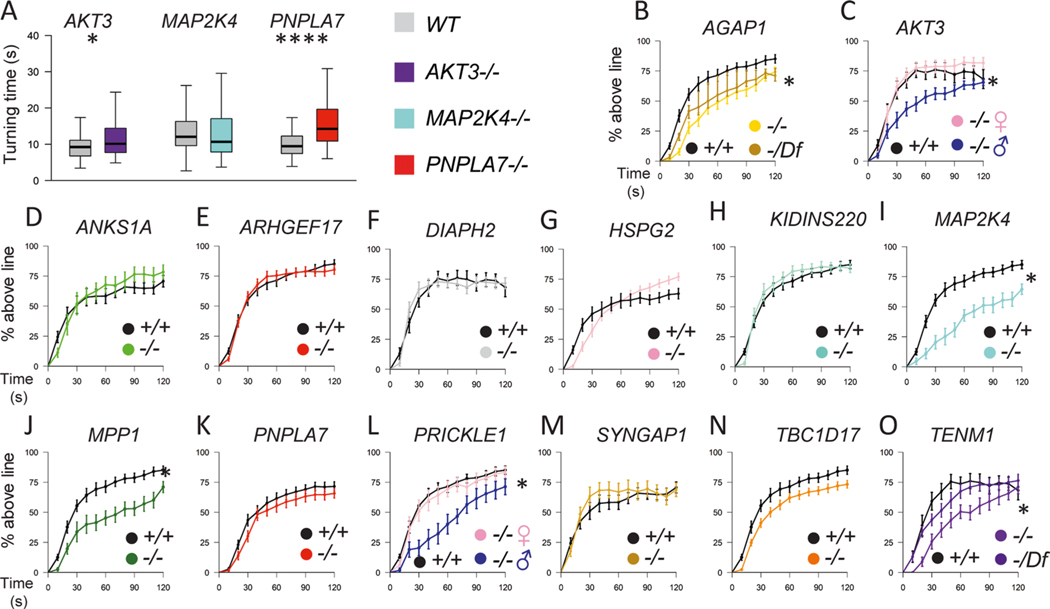
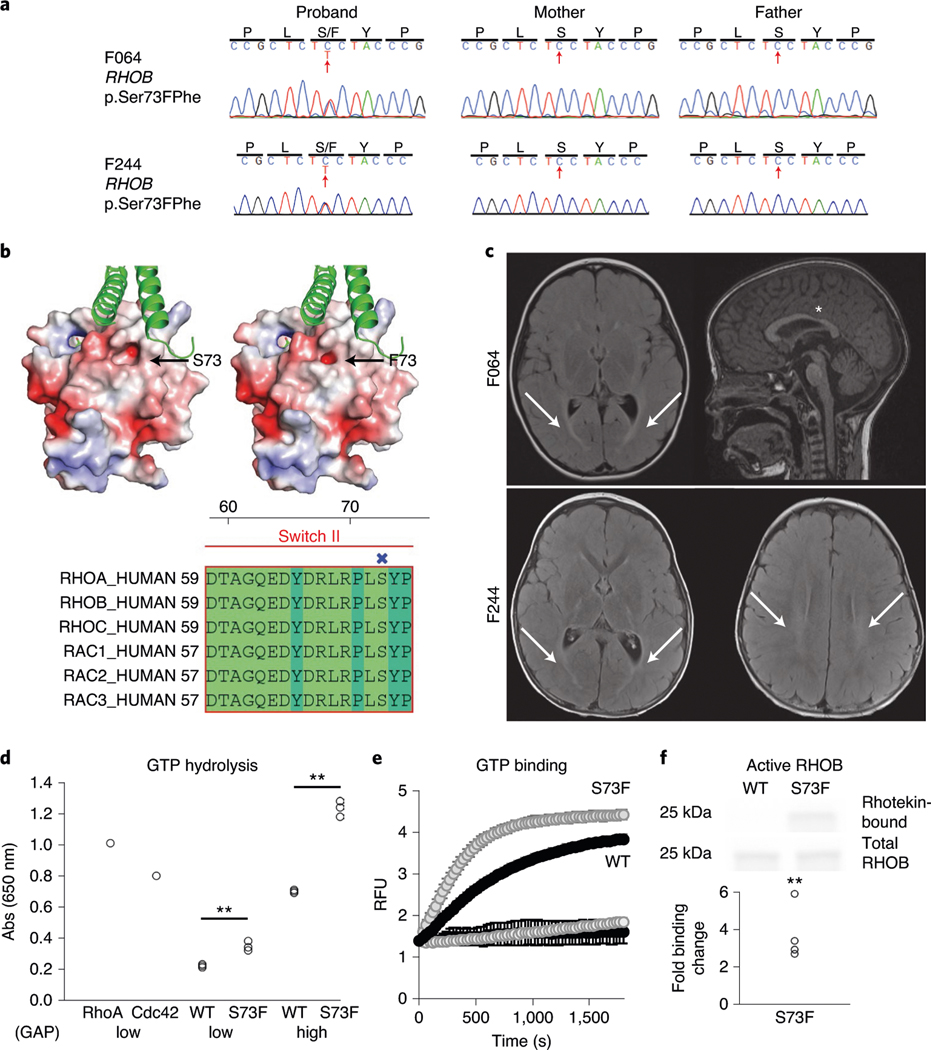
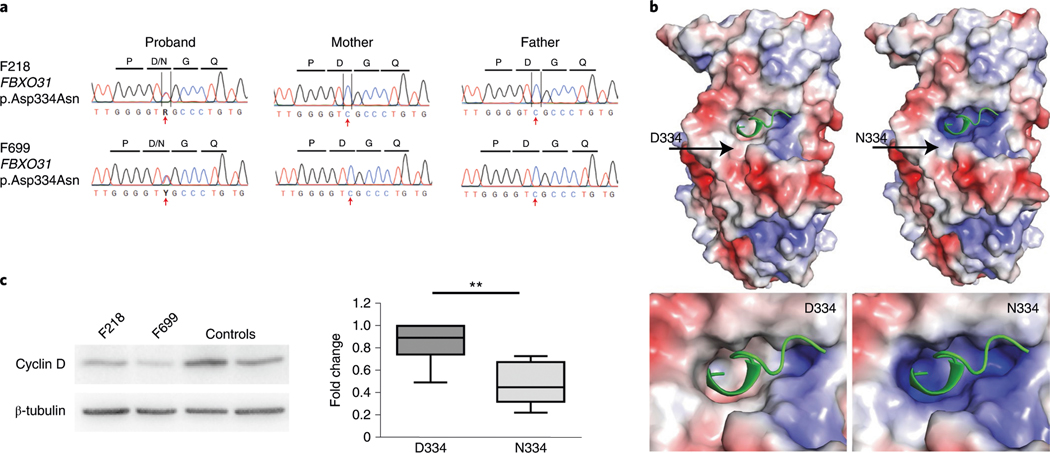
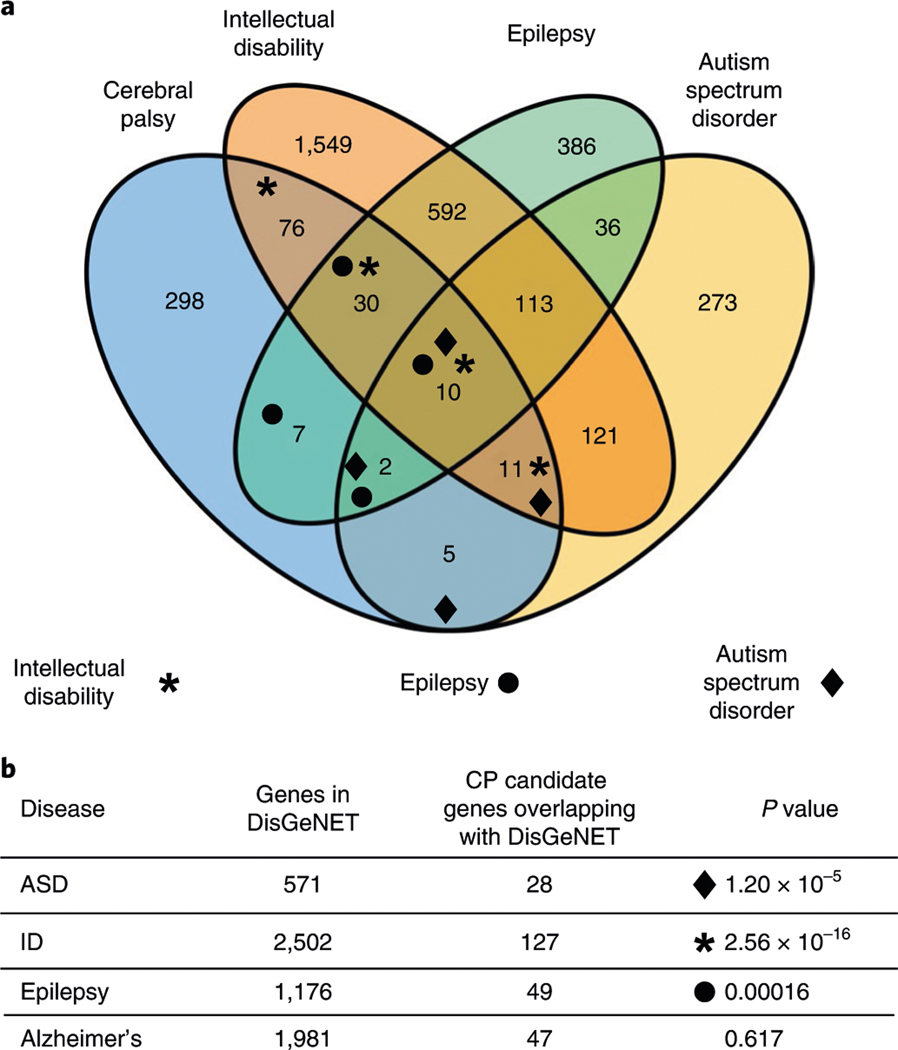
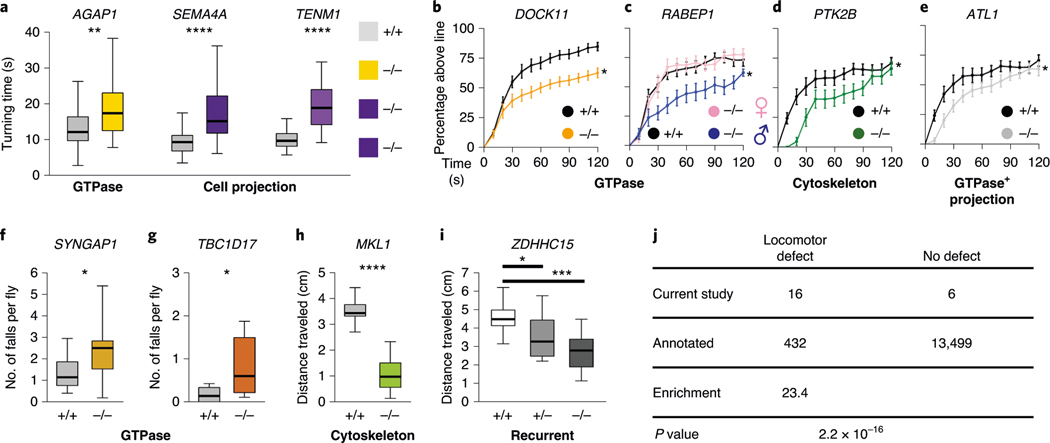
In addition to commonly associated environmental factors, genomic factors may cause cerebral palsy. We performed whole-exome sequencing of 250 parent-offspring trios, and observed enrichment of damaging de novo mutations in cerebral palsy cases. Eight genes had multiple damaging de novo mutations; of these, two (TUBA1A and CTNNB1) met genome-wide significance. We identified two novel monogenic etiologies, FBXO31 and RHOB, and showed that the RHOB mutation enhances active-state Rho effector binding while the FBXO31 mutation diminishes cyclin D levels. Candidate cerebral palsy risk genes overlapped with neurodevelopmental disorder genes. Network analyses identified enrichment of Rho GTPase, extracellular matrix, focal adhesion and cytoskeleton pathways. Cerebral palsy risk genes in enriched pathways were shown to regulate neuromotor function in a Drosophila reverse genetics screen. We estimate that 14% of cases could be attributed to an excess of damaging de novo or recessive variants. These findings provide evidence for genetically mediated dysregulation of early neuronal connectivity in cerebral palsy.
Figures

References
-
- Oskoui M, Coutinho F, Dykeman J, Jette N & Pringsheim T An update on the prevalence of cerebral palsy: a systematic review and meta-analysis. Dev. Med. Child Neurol. 55, 509–519 (2013). - PubMed
-
- Cans C Surveillance of cerebral palsy in Europe: a collaboration of cerebral palsy surveys and registers. Dev. Med. Child Neurol. 42, 816–824 (2000). - PubMed
-
- Longo LD & Ashwal S William Osler, Sigmund Freud and the evolution of ideas concerning cerebral palsy. J. Hist. Neurosci. 2, 255–282 (1993). - PubMed
-
- Panteliadis C, Panteliadis P & Vassilyadi F Hallmarks in the history of cerebral palsy: from antiquity to mid-20th century. Brain Dev. 35, 285–292 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
