

Leveraging Computational Modeling to Understand Infectious Diseases
- PMID: 32989410
- PMCID: PMC7511257
- DOI: 10.1007/s40139-020-00213-x
Leveraging Computational Modeling to Understand Infectious Diseases
Abstract
Purpose of review: Computational and mathematical modeling have become a critical part of understanding in-host infectious disease dynamics and predicting effective treatments. In this review, we discuss recent findings pertaining to the biological mechanisms underlying infectious diseases, including etiology, pathogenesis, and the cellular interactions with infectious agents. We present advances in modeling techniques that have led to fundamental disease discoveries and impacted clinical translation.
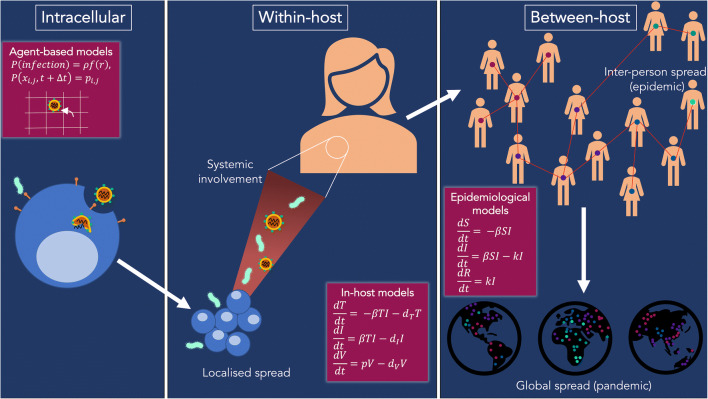
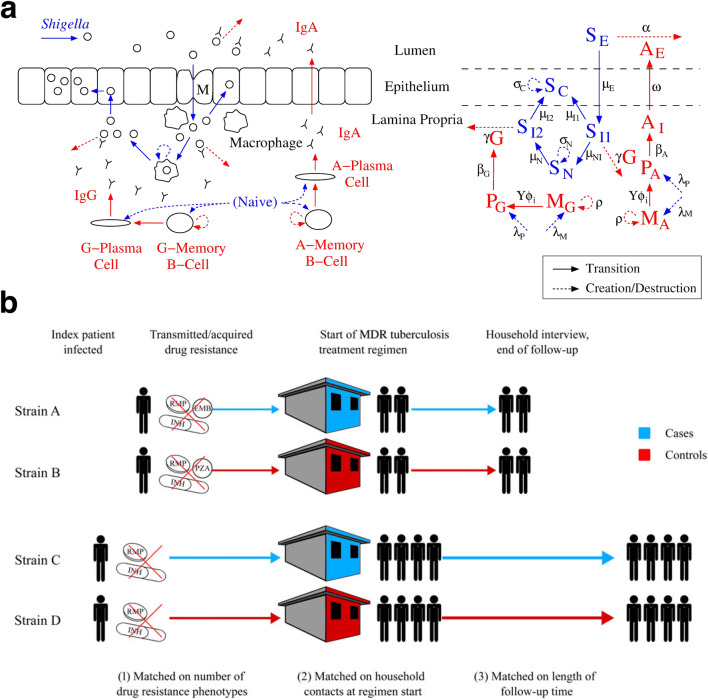
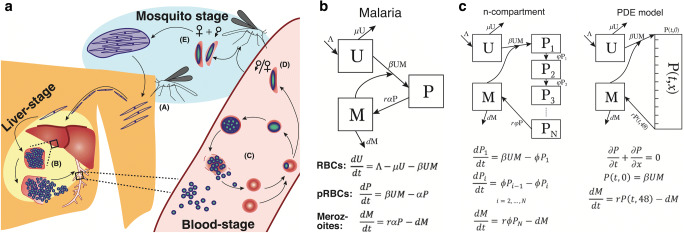
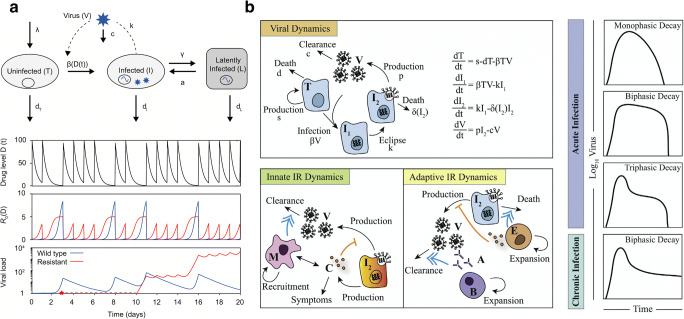
Recent findings: Combining mechanistic models and machine learning algorithms has led to improvements in the treatment of Shigella and tuberculosis through the development of novel compounds. Modeling of the epidemic dynamics of malaria at the within-host and between-host level has afforded the development of more effective vaccination and antimalarial therapies. Similarly, in-host and host-host models have supported the development of new HIV treatment modalities and an improved understanding of the immune involvement in influenza. In addition, large-scale transmission models of SARS-CoV-2 have furthered the understanding of coronavirus disease and allowed for rapid policy implementations on travel restrictions and contract tracing apps.
Summary: Computational modeling is now more than ever at the forefront of infectious disease research due to the COVID-19 pandemic. This review highlights how infectious diseases can be better understood by connecting scientists from medicine and molecular biology with those in computer science and applied mathematics.
Keywords: Bacteria; Computational modeling; Infectious diseases; Mathematics; Parasites; Viruses.
© Springer Science+Business Media, LLC, part of Springer Nature 2020.
Conflict of interest statement
Conflict of InterestAdrianne L. Jenner, Rosemary A. Aogo, Courtney L. Davis, Amber M. Smith, and Morgan Craig declare that they have no conflict of interest.
Figures
References
-
- Fenton A. Editorial: mathematical modelling of infectious diseases. Parasitology. 2016;143:801–804. - PubMed
-
- Khoury DS, Aogo R, Randriafanomezantsoa-Radohery G, McCaw JM, Simpson JA, McCarthy JS, Haque A, Cromer D, Davenport MP. Within-host modeling of blood-stage malaria. Immunol Rev. 2018;285:168–193. - PubMed
-
- Dodd PJ, Sismanidis C, Seddon JA. Global burden of drug-resistant tuberculosis in children: a mathematical modelling study. Lancet Infect Dis. 2016;16:1193–1201. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous