

Genome-wide CRISPR screens of oral squamous cell carcinoma reveal fitness genes in the Hippo pathway
- PMID: 32990596
- PMCID: PMC7591259
- DOI: 10.7554/eLife.57761
Genome-wide CRISPR screens of oral squamous cell carcinoma reveal fitness genes in the Hippo pathway
Abstract
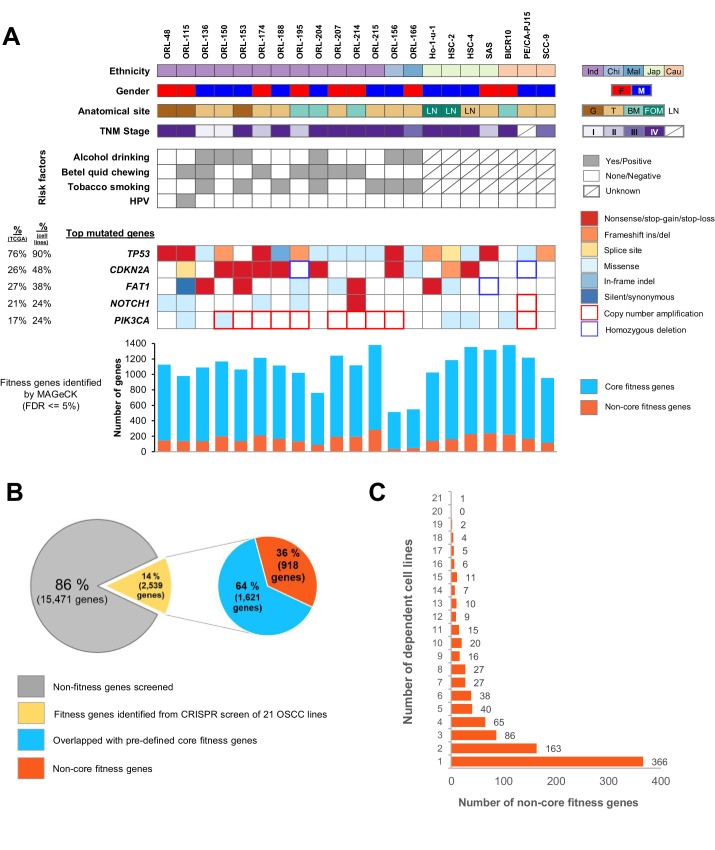
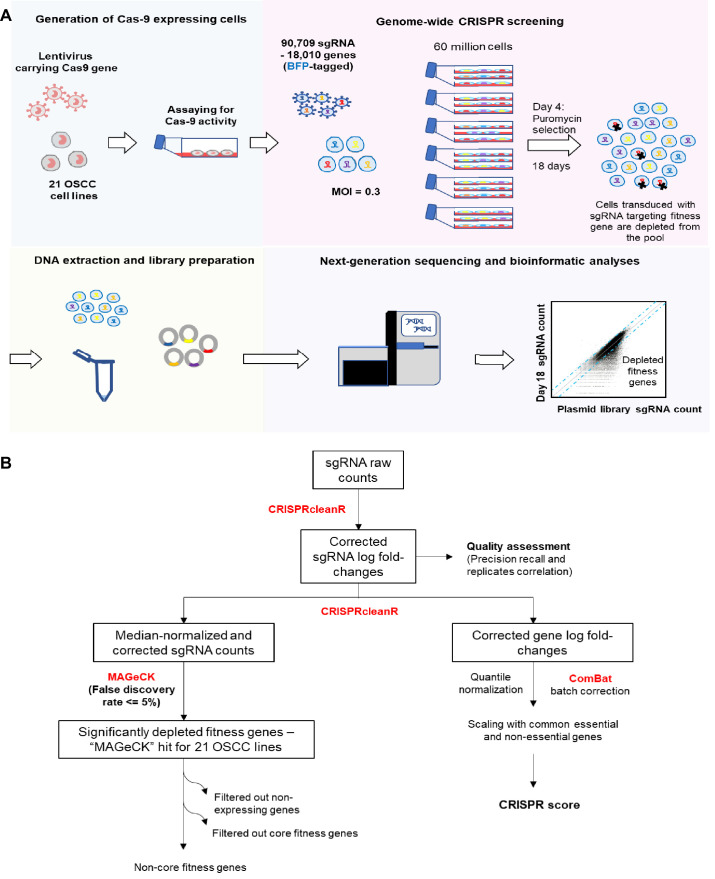
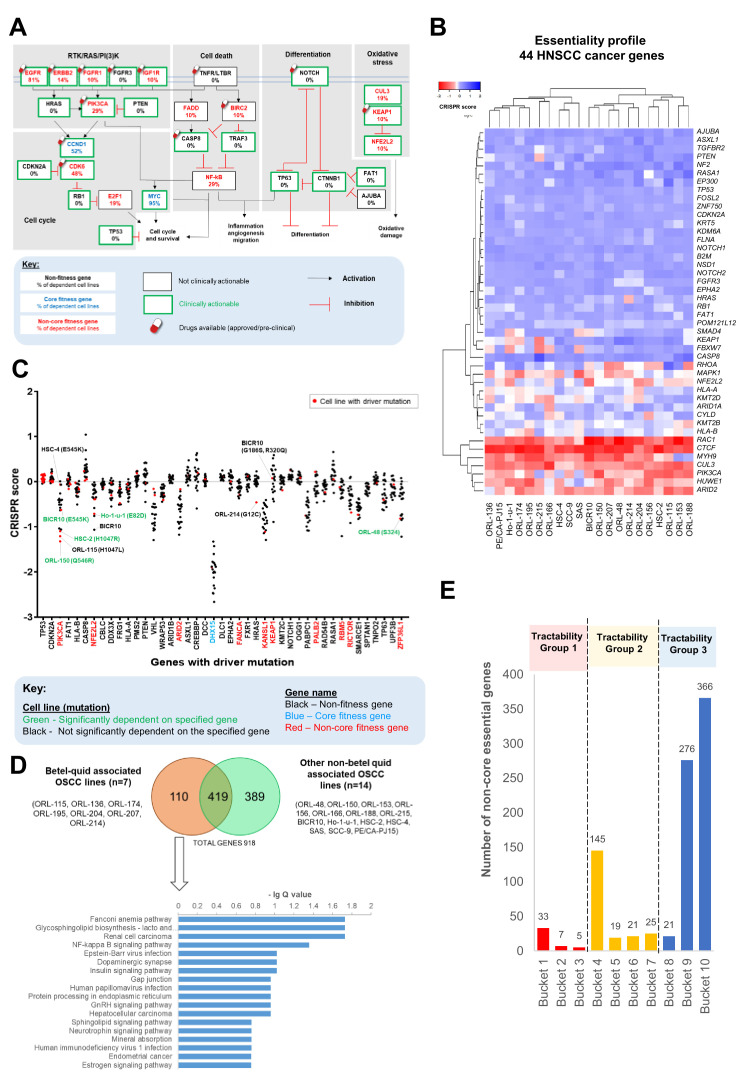
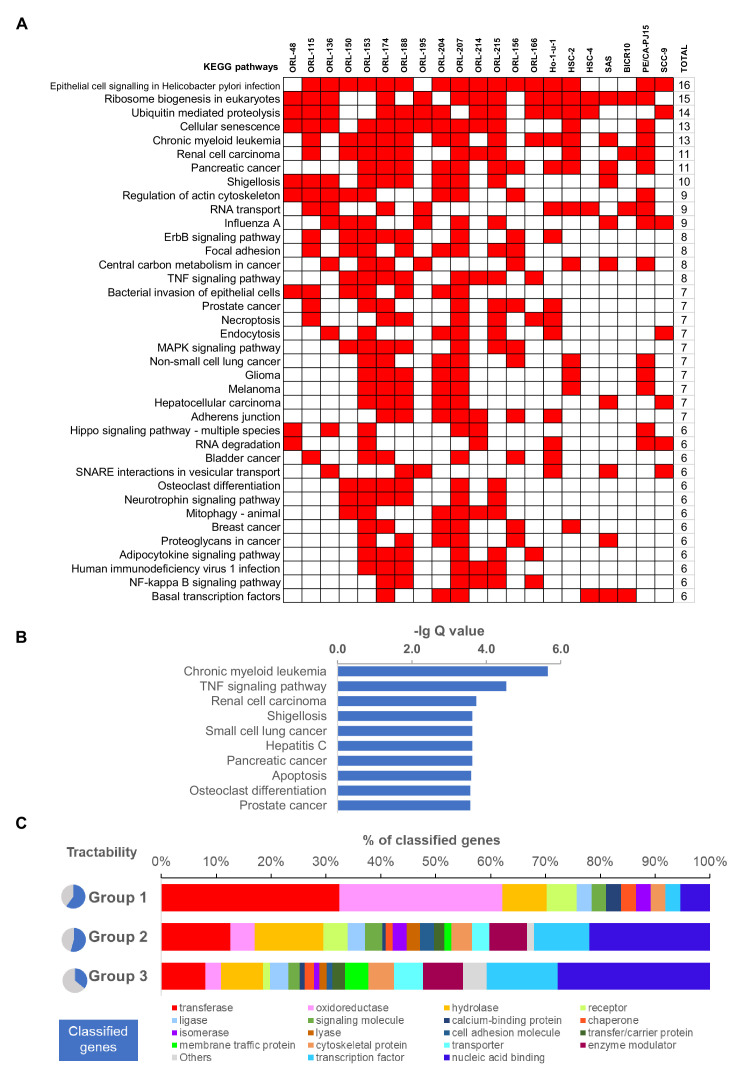
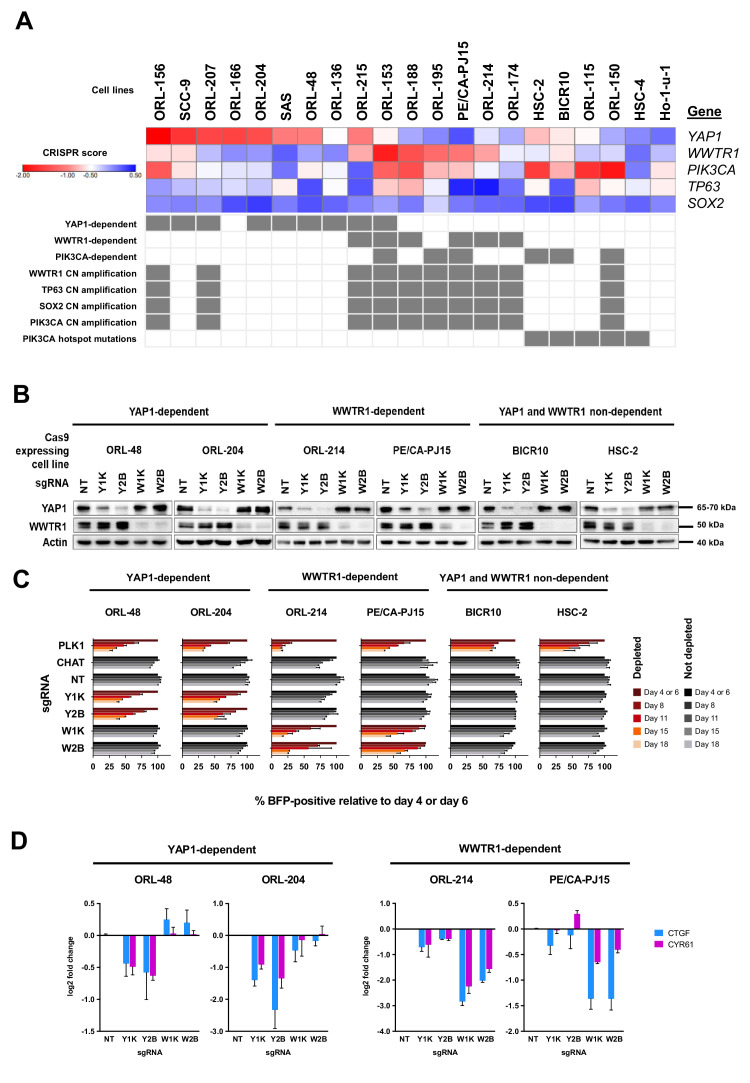
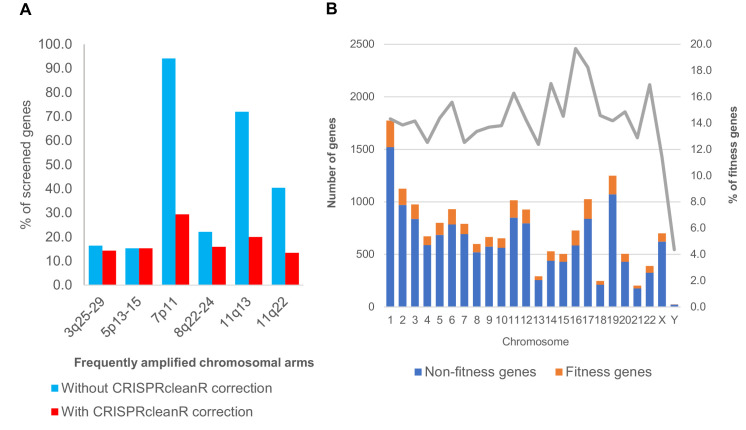
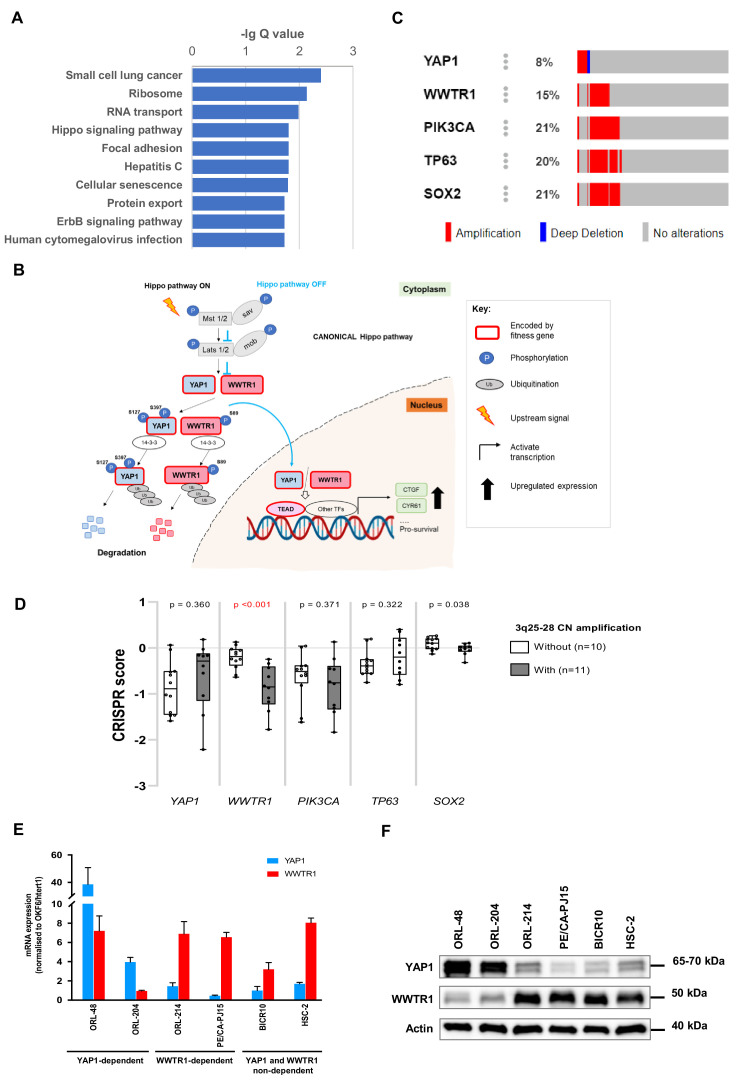
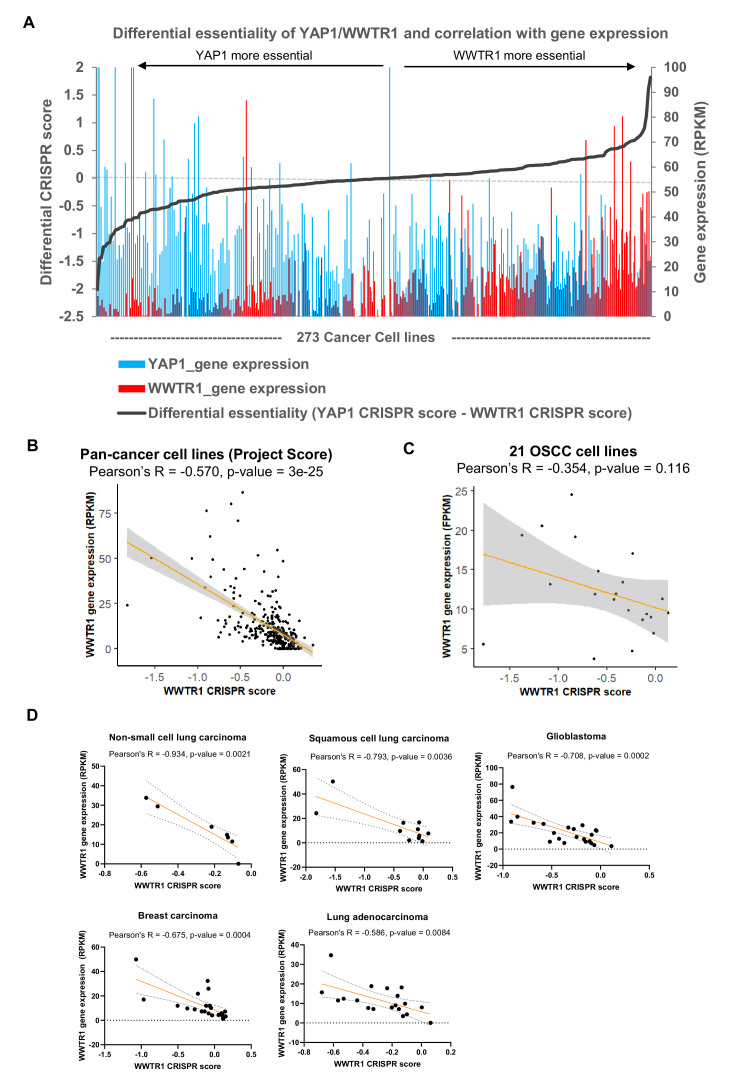
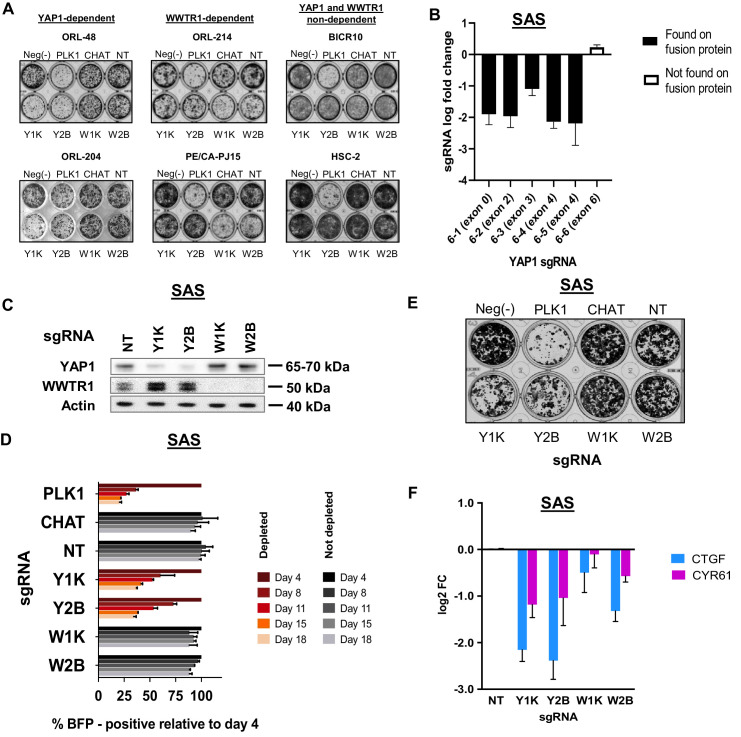
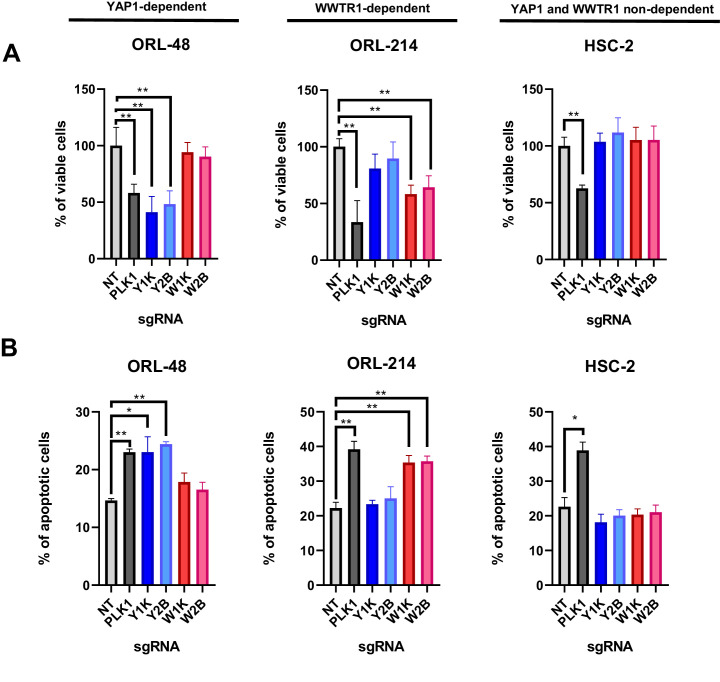
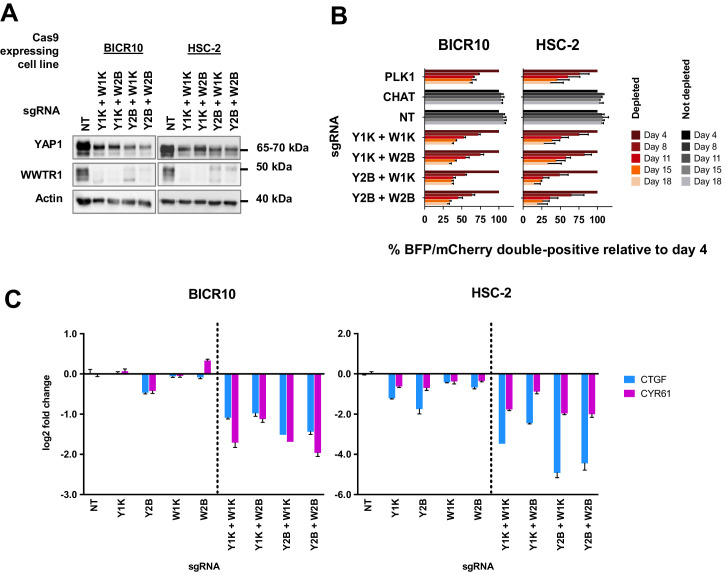
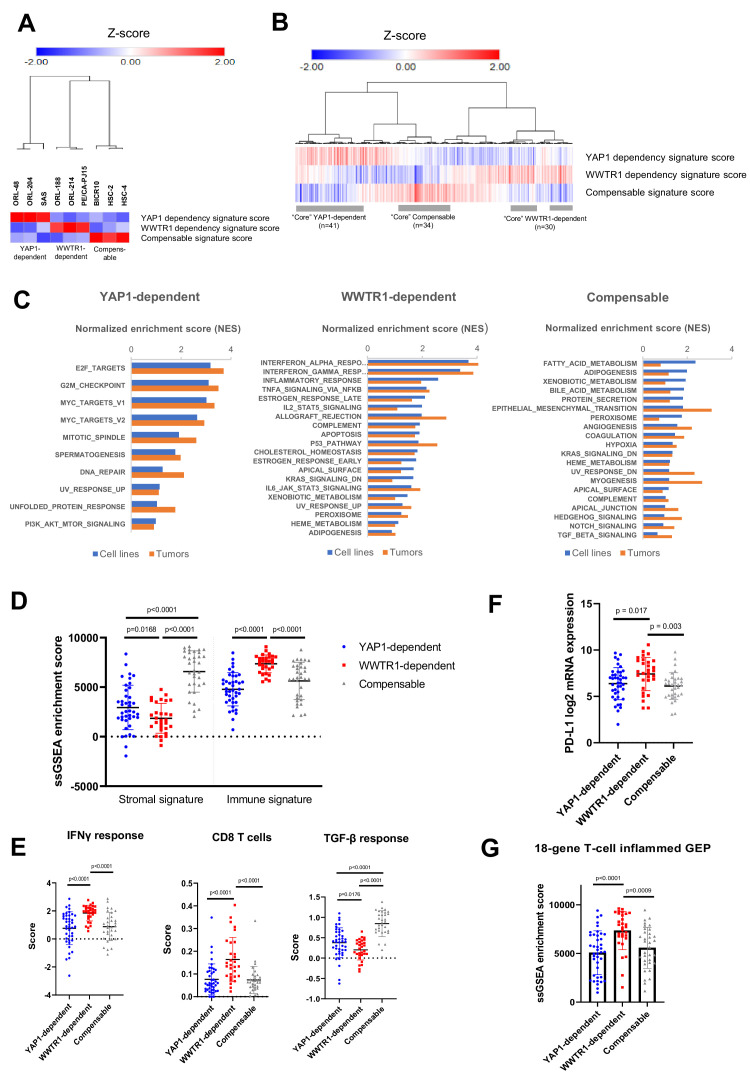
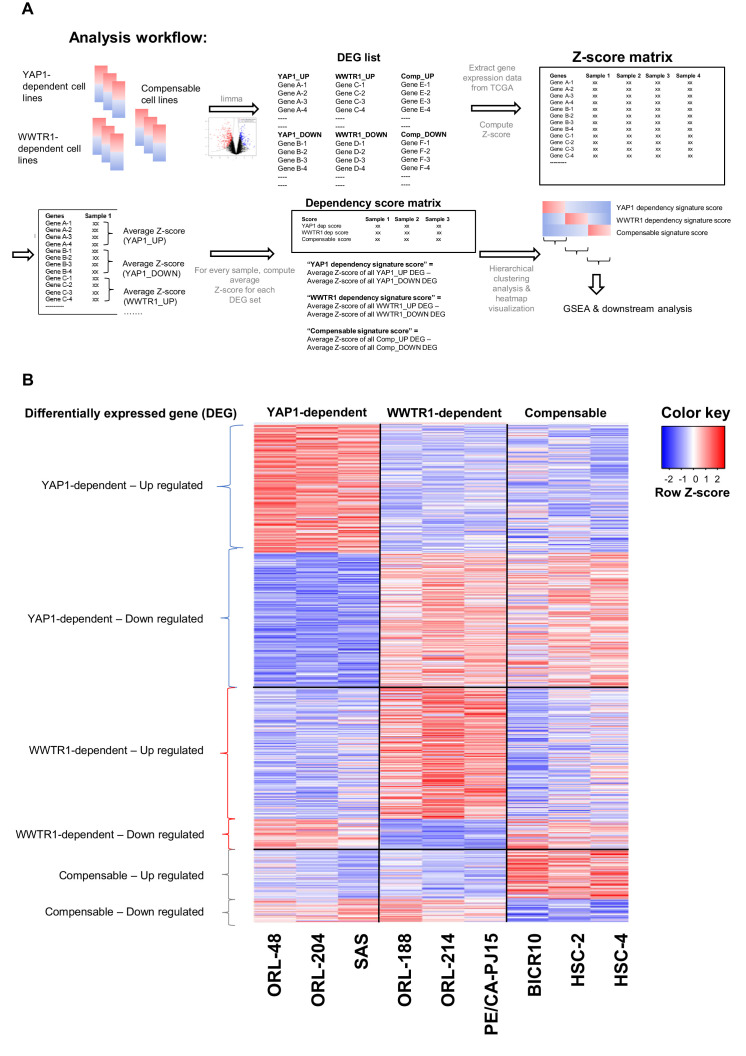
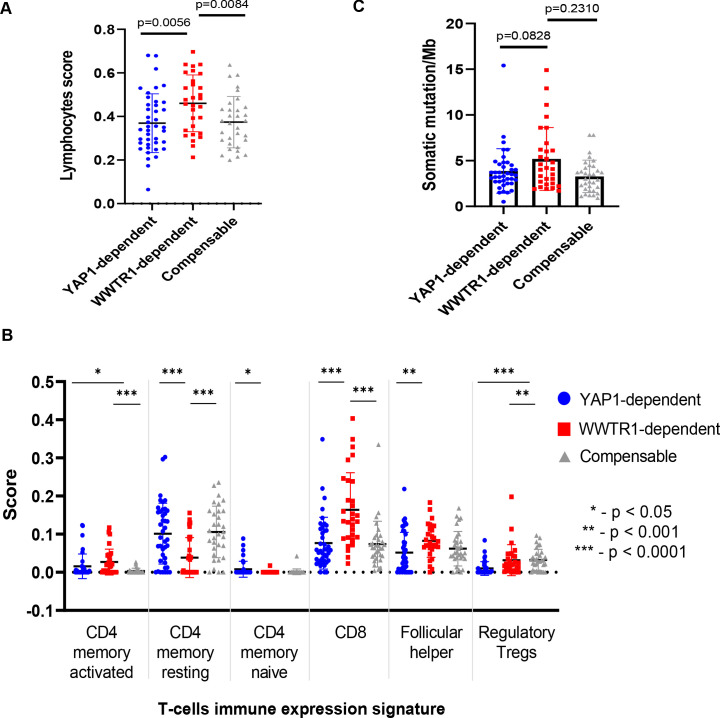
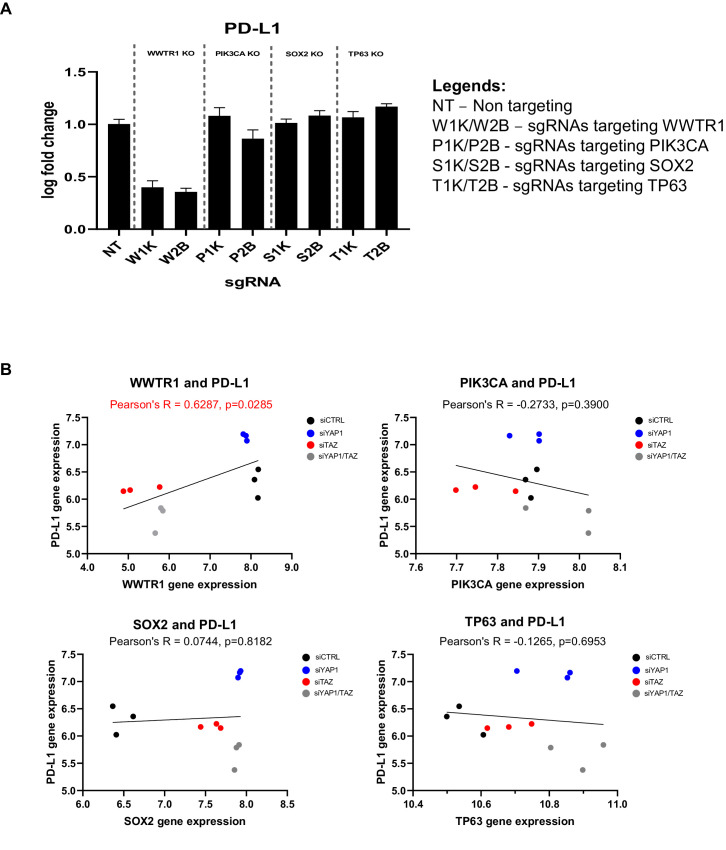
New therapeutic targets for oral squamous cell carcinoma (OSCC) are urgently needed. We conducted genome-wide CRISPR-Cas9 screens in 21 OSCC cell lines, primarily derived from Asians, to identify genetic vulnerabilities that can be explored as therapeutic targets. We identify known and novel fitness genes and demonstrate that many previously identified OSCC-related cancer genes are non-essential and could have limited therapeutic value, while other fitness genes warrant further investigation for their potential as therapeutic targets. We validate a distinctive dependency on YAP1 and WWTR1 of the Hippo pathway, where the lost-of-fitness effect of one paralog can be compensated only in a subset of lines. We also discover that OSCCs with WWTR1 dependency signature are significantly associated with biomarkers of favorable response toward immunotherapy. In summary, we have delineated the genetic vulnerabilities of OSCC, enabling the prioritization of therapeutic targets for further exploration, including the targeting of YAP1 and WWTR1.
Keywords: CRISPR screen; Hippo pathway; cancer biology; fitness genes; genetics; genomics; human; oral squamous cell carcinoma; therapeutic targets.
Plain language summary
Many types of cancer now have 'targeted treatments', which specifically home in on genes cancer cells rely on for survival. But there are very few of these treatments available for the most common type of mouth cancer, oral squamous cell carcinoma, which around 350,000 people are diagnosed with each year. Designing targeted treatments relies on detailed knowledge of the genetic makeup of the cancer cells. But, little is known about which genes drive oral squamous cell carcinoma, especially among patients living in Asia, which is where over half of yearly cases are diagnosed. One way to resolve this is to use gene editing technology to find the genes that the cancer cells need to survive. Now, Chai et al. have used a gene editing tool known as CRISPR to examine 21 cell lines from patients diagnosed with oral squamous cell carcinoma. Most of these lines were from Asian patients, some of whom had a history of chewing betel quid which increases the risk of mouth cancer. By individually inactivating genes in these cell lines one by one, Chai et al. were able to identify 918 genes linked to the survival of the cancer cells. Some of these genes have already been associated with the spread of other types of cancer, whereas others are completely unique to oral squamous cell carcinoma. The screen also discovered that some cell lines could not survive without genes involved in a signalling pathway called Hippo, which is known to contribute to the progression of many other types of cancer. Uncovering the genes associated with oral squamous cell carcinoma opens the way for the development of new targeted treatments. Targeted therapies already exist for some of the genes identified in this study, and it may be possible to repurpose them as a treatment for this widespread mouth cancer. But, given that different cell lines relied on different genes to survive, the next step will be to identify which genes to inactivate in each patient.
© 2020, Chai et al.
Conflict of interest statement
AC, PY, SP, SY, HL, VT, EG, FB, JB, JG, AT, MG, SC No competing interests declared, UM is affiliated with AstraZeneca
Figures
References
-
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA, Yearley J, Seiwert TY, Ribas A, McClanahan TK. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. Journal of Clinical Investigation. 2017;127:2930–2940. doi: 10.1172/JCI91190. - DOI - PMC - PubMed
-
- Barazas M, Annunziato S, Pettitt SJ, de Krijger I, Ghezraoui H, Roobol SJ, Lutz C, Frankum J, Song FF, Brough R, Evers B, Gogola E, Bhin J, van de Ven M, van Gent DC, Jacobs JJL, Chapman R, Lord CJ, Jonkers J, Rottenberg S. The CST complex mediates end protection at Double-Strand breaks and promotes PARP inhibitor sensitivity in BRCA1-Deficient cells. Cell Reports. 2018;23:2107–2118. doi: 10.1016/j.celrep.2018.04.046. - DOI - PMC - PubMed
-
- Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, Saba NF, Weiss J, Wirth L, Sukari A, Kang H, Gibson MK, Massarelli E, Powell S, Meister A, Shu X, Cheng JD, Haddad R. Pembrolizumab for platinum- and Cetuximab-Refractory head and neck Cancer: results from a Single-Arm, phase II study. Journal of Clinical Oncology. 2017;35:1542–1549. doi: 10.1200/JCO.2016.70.1524. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
