16p11.2 Copy Number Variations and Neurodevelopmental Disorders
- PMID: 32993859
- PMCID: PMC7606557
- DOI: 10.1016/j.tins.2020.09.001
16p11.2 Copy Number Variations and Neurodevelopmental Disorders
Abstract
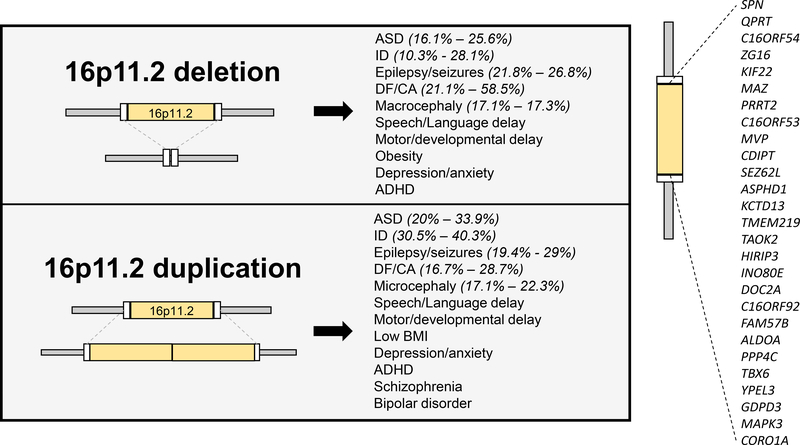
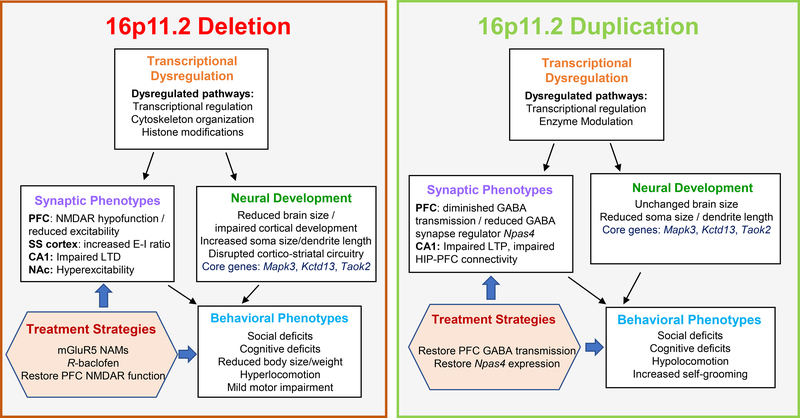
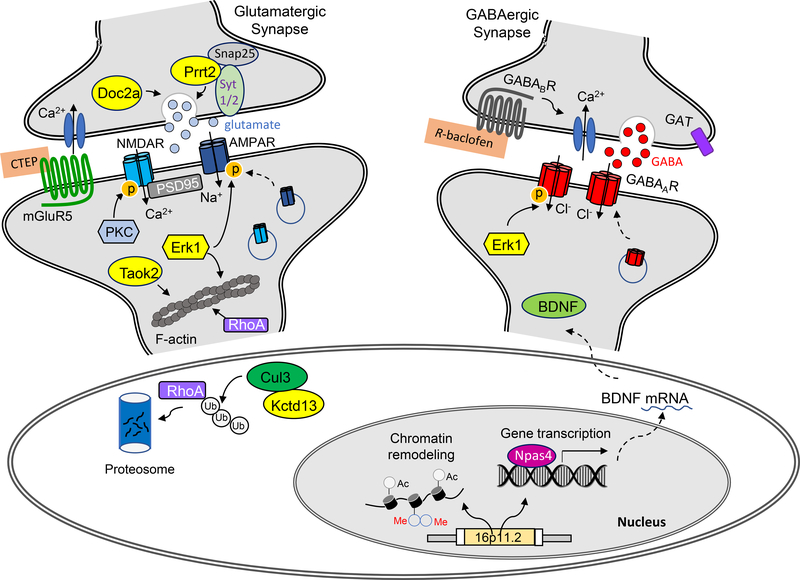
Copy number variations (CNVs) of the human 16p11.2 genetic locus are associated with a range of neurodevelopmental disorders, including autism spectrum disorder, intellectual disability, and epilepsy. In this review, we delineate genetic information and diverse phenotypes in individuals with 16p11.2 CNVs, and synthesize preclinical findings from transgenic mouse models of 16p11.2 CNVs. Mice with 16p11.2 deletions or duplications recapitulate many core behavioral phenotypes, including social and cognitive deficits, and exhibit altered synaptic function across various brain areas. Mechanisms of transcriptional dysregulation and cortical maldevelopment are reviewed, along with potential therapeutic intervention strategies.
Keywords: 16p11.2 deletion and duplication; autism spectrum disorders; clinical phenotypes; mouse models; prefrontal cortex.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Conflict of interest statement
CONFLICT OF INTEREST
The authors declare no conflict of interest related to this work (financial or else).
Figures