

Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity
- PMID: 32994364
- PMCID: PMC7857405
- DOI: 10.1126/science.abd4250
Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity
Abstract
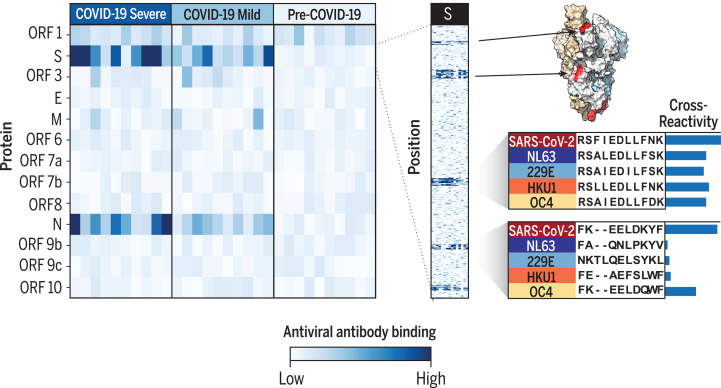
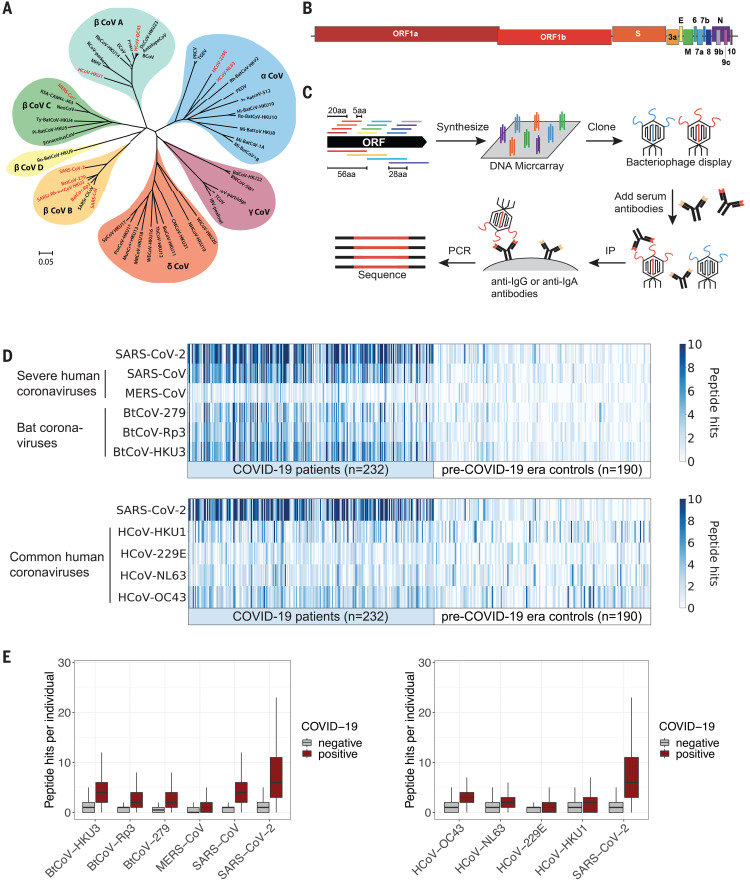
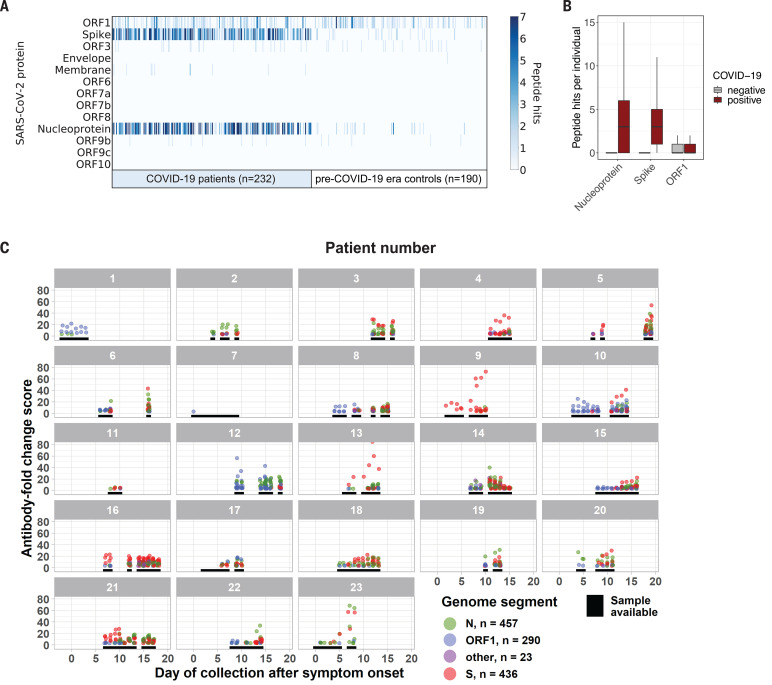
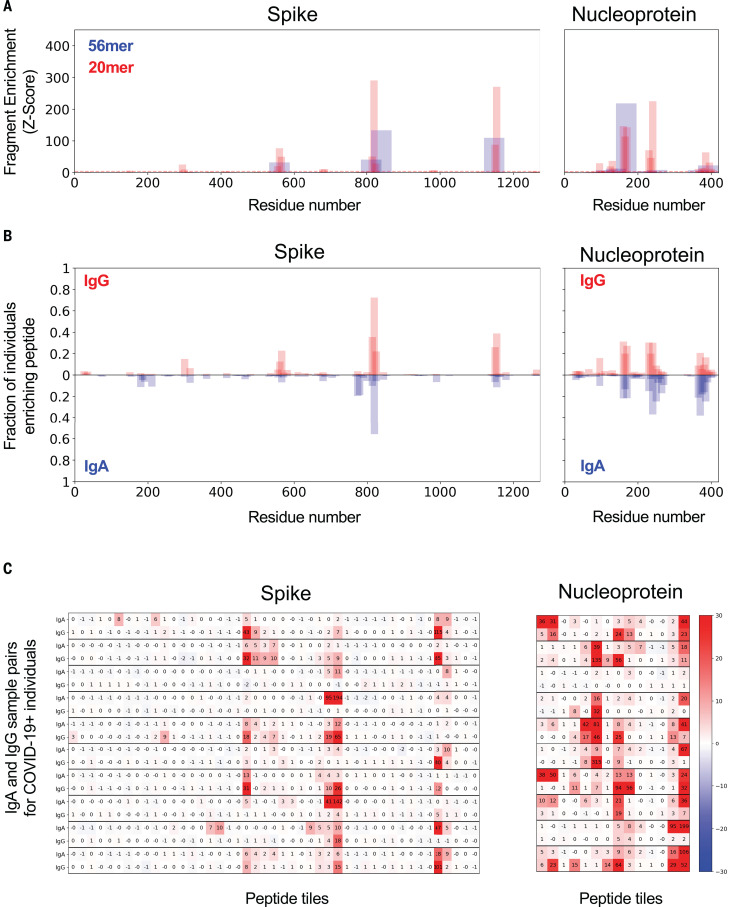
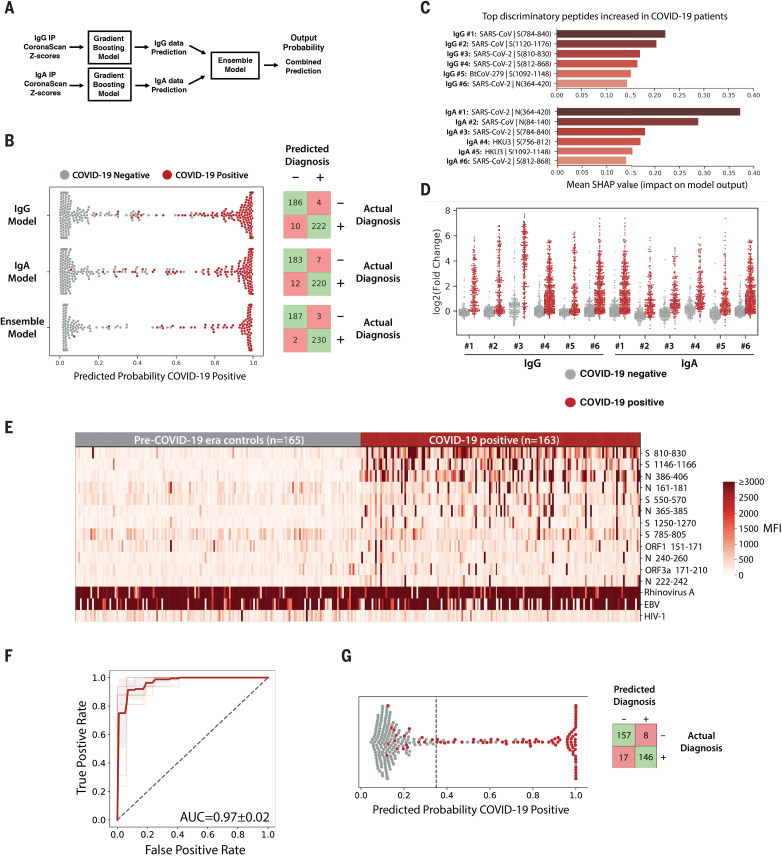
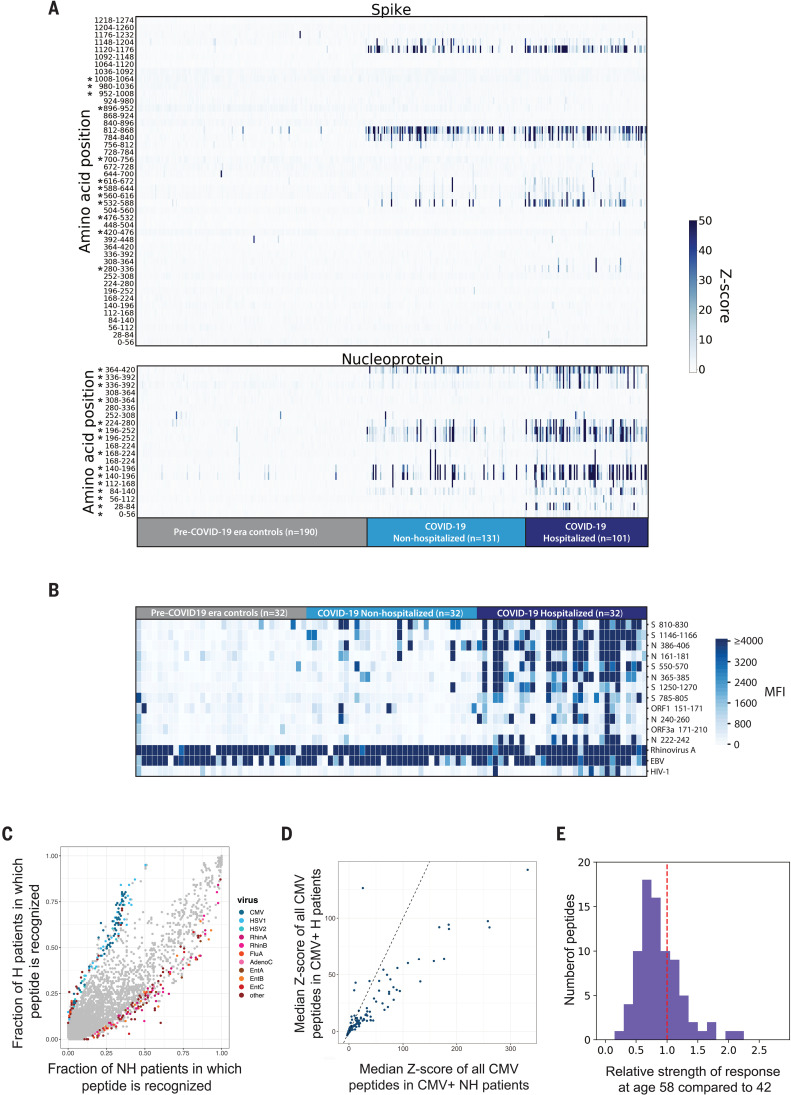
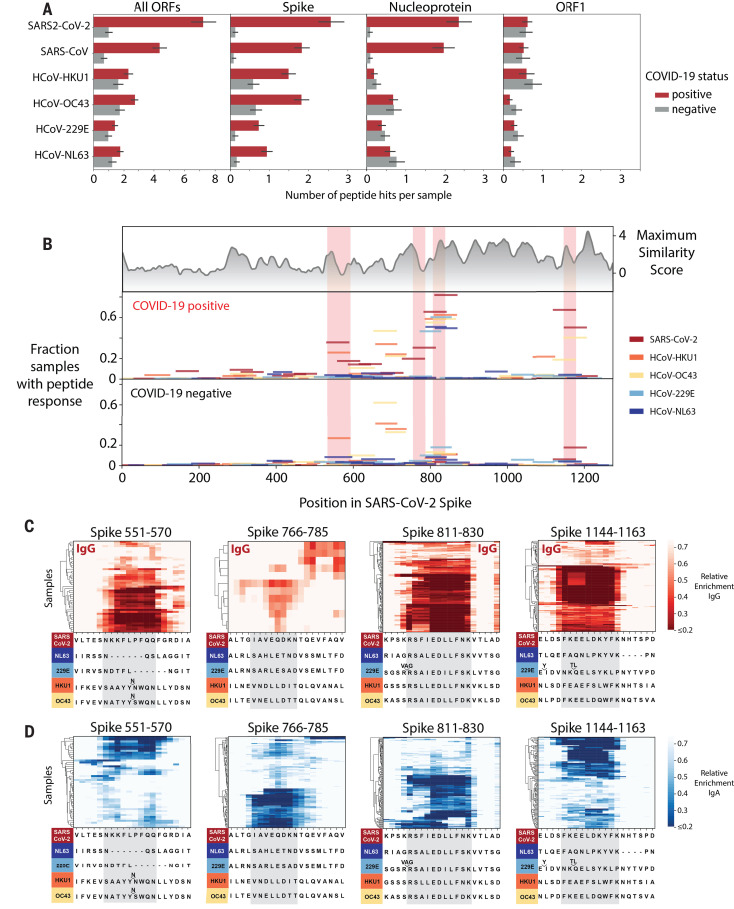
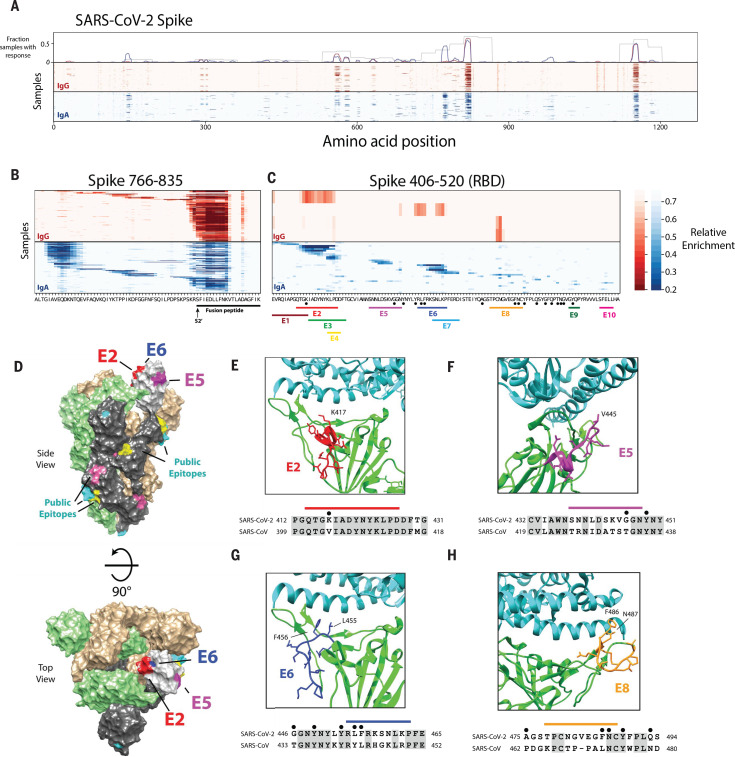
Understanding humoral responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is critical for improving diagnostics, therapeutics, and vaccines. Deep serological profiling of 232 coronavirus disease 2019 (COVID-19) patients and 190 pre-COVID-19 era controls using VirScan revealed more than 800 epitopes in the SARS-CoV-2 proteome, including 10 epitopes likely recognized by neutralizing antibodies. Preexisting antibodies in controls recognized SARS-CoV-2 ORF1, whereas only COVID-19 patient antibodies primarily recognized spike protein and nucleoprotein. A machine learning model trained on VirScan data predicted SARS-CoV-2 exposure history with 99% sensitivity and 98% specificity; a rapid Luminex-based diagnostic was developed from the most discriminatory SARS-CoV-2 peptides. Individuals with more severe COVID-19 exhibited stronger and broader SARS-CoV-2 responses, weaker antibody responses to prior infections, and higher incidence of cytomegalovirus and herpes simplex virus 1, possibly influenced by demographic covariates. Among hospitalized patients, males produce stronger SARS-CoV-2 antibody responses than females.
Copyright © 2020, American Association for the Advancement of Science.
Figures
Comment in
-
COVID-19: cross-immunity of viral epitopes may influence severity of infection and immune response.Signal Transduct Target Ther. 2021 Mar 1;6(1):102. doi: 10.1038/s41392-021-00490-x. Signal Transduct Target Ther. 2021. PMID: 33649300 Free PMC article. No abstract available.
References
-
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU); https://coronavirus.jhu.edu/map.html.
Publication types
MeSH terms
Substances
Grants and funding
- R01 AI139538/AI/NIAID NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- R01 AI148232/AI/NIAID NIH HHS/United States
- U24 AI118633/AI/NIAID NIH HHS/United States
- K08 DK114563/DK/NIDDK NIH HHS/United States
- 201387/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- U01 AI138318/AI/NIAID NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- T32 AI007044/AI/NIAID NIH HHS/United States
- P30 AR070253/AR/NIAMS NIH HHS/United States
- R01 AI092905/AI/NIAID NIH HHS/United States
- R01 AI121394/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
