

Genetic inactivation of RIP1 kinase does not ameliorate disease in a mouse model of ALS
- PMID: 32994544
- PMCID: PMC7937687
- DOI: 10.1038/s41418-020-00625-7
Genetic inactivation of RIP1 kinase does not ameliorate disease in a mouse model of ALS
Abstract
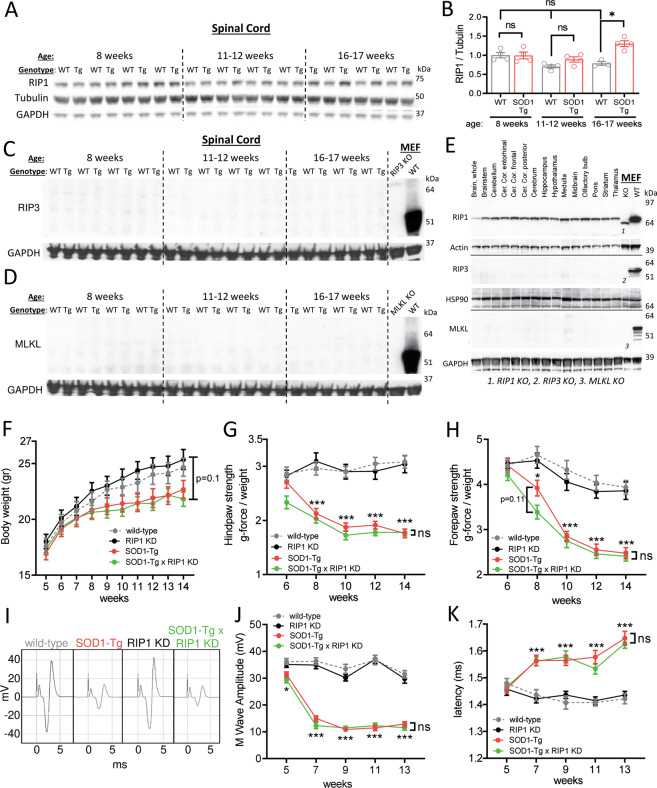
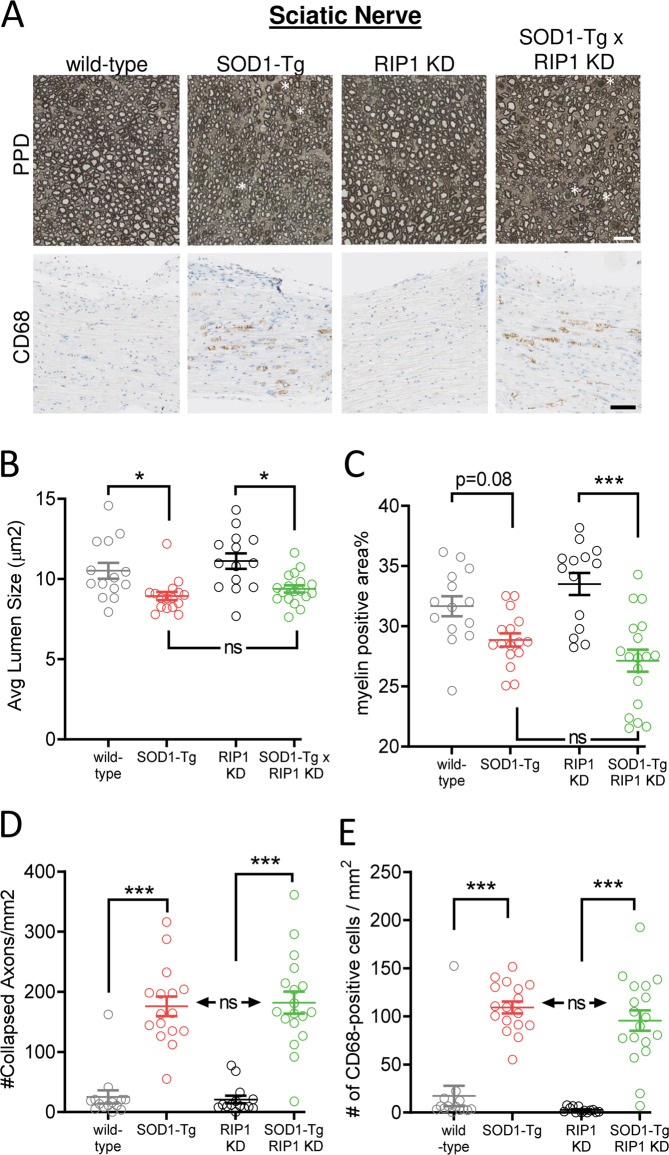
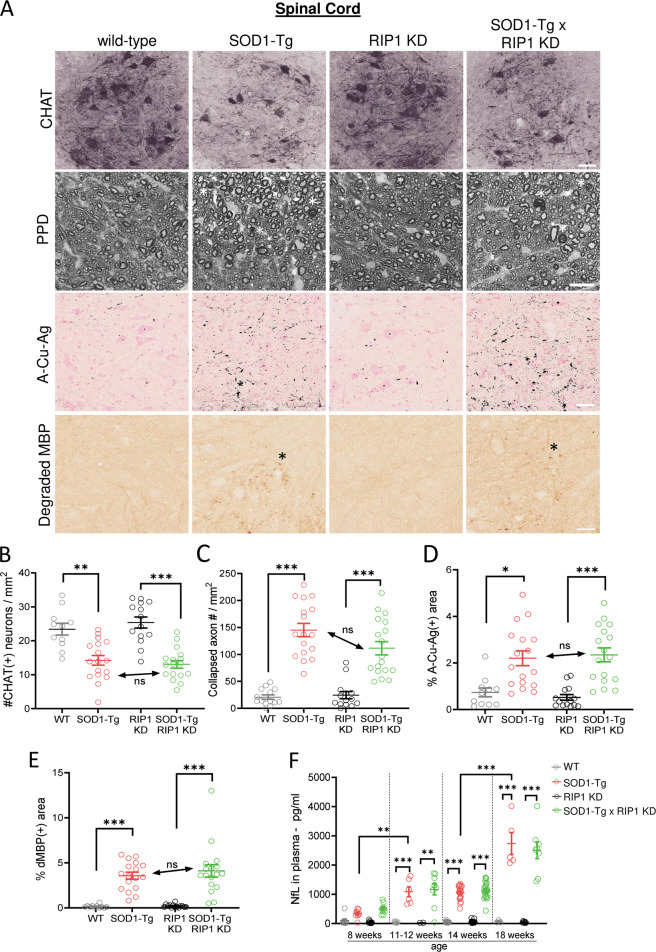
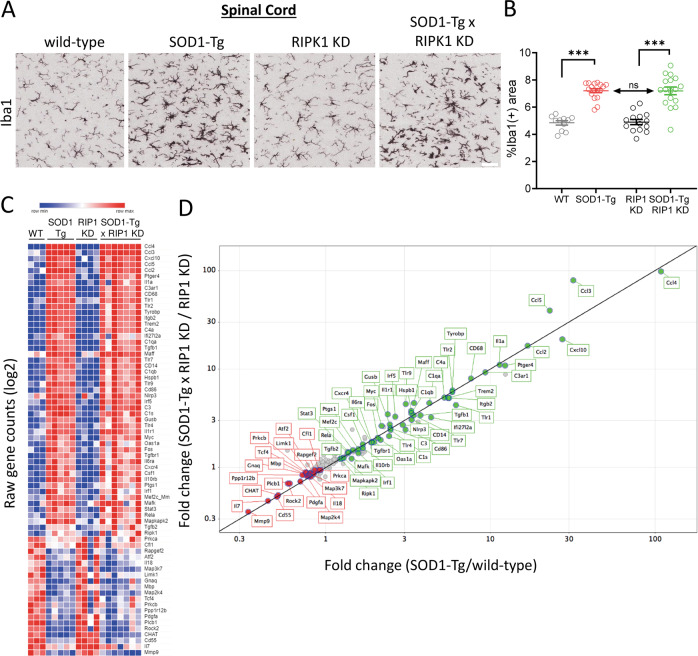
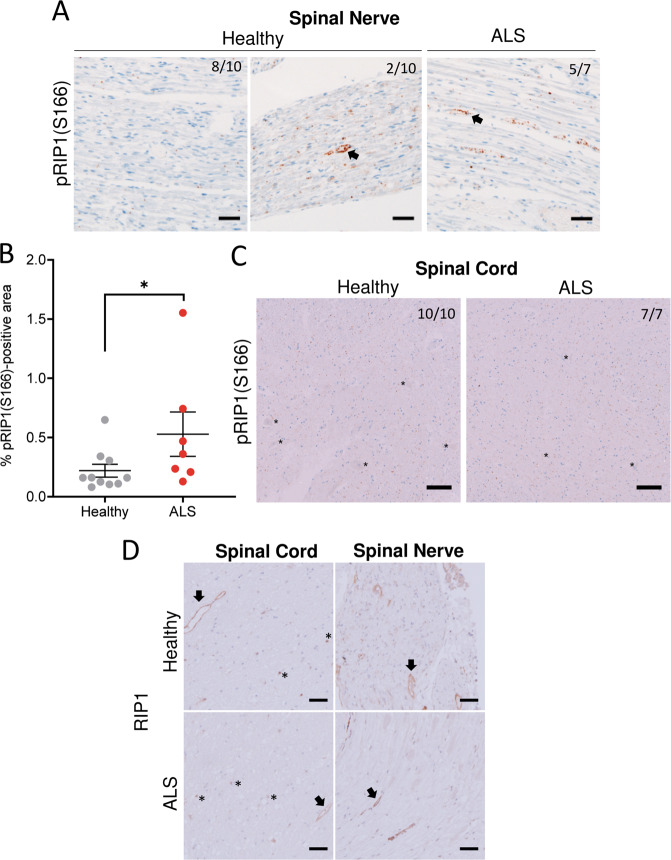
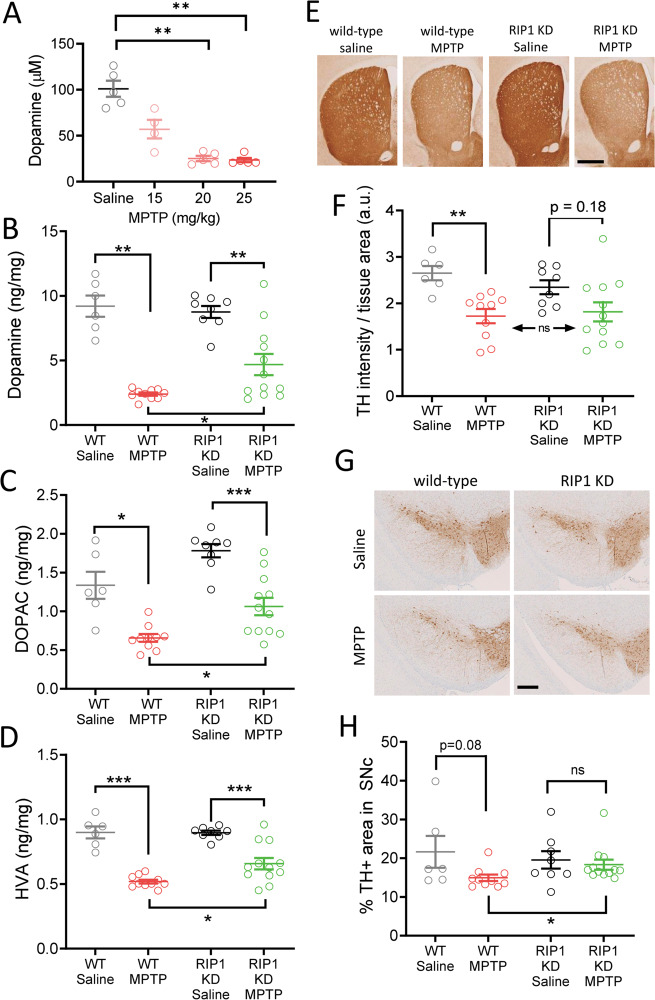
RIP1 kinase is proposed to play a critical role in driving necroptosis and inflammation in neurodegenerative disorders, including Amyotrophic Lateral Sclerosis (ALS). Preclinical studies indicated that while pharmacological inhibition of RIP1 kinase can ameliorate axonal pathology and delay disease onset in the mutant SOD1 transgenic (SOD1-Tg) mice, genetic blockade of necroptosis does not provide benefit in this mouse model. To clarify the role of RIP1 kinase activity in driving pathology in SOD1-Tg mice, we crossed SOD1-Tgs to RIP1 kinase-dead knock-in mice, and measured disease progression using functional and histopathological endpoints. Genetic inactivation of the RIP1 kinase activity in the SOD1-Tgs did not benefit the declining muscle strength or nerve function, motor neuron degeneration or neuroinflammation. In addition, we did not find evidence of phosphorylated RIP1 accumulation in the spinal cords of ALS patients. On the other hand, genetic inactivation of RIP1 kinase activity ameliorated the depletion of the neurotransmitter dopamine in a toxin model of dopaminergic neurodegeneration. These findings indicate that RIP1 kinase activity is dispensable for disease pathogenesis in the SOD1-Tg mice while inhibition of kinase activity may provide benefit in acute injury models.
Conflict of interest statement
TE and BB are former and all other authors are current employees of Genentech, Inc.
Figures
References
-
- Martens S, Hofmans S, Declercq W, Augustyns K, Vandenabeele P. Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci. 2020;41:209–24. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
