

This is a preprint.
Functional genomic screens identify human host factors for SARS-CoV-2 and common cold coronaviruses
- PMID: 32995787
- PMCID: PMC7523113
- DOI: 10.1101/2020.09.24.312298
Functional genomic screens identify human host factors for SARS-CoV-2 and common cold coronaviruses
Update in
-
Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses.Cell. 2021 Jan 7;184(1):106-119.e14. doi: 10.1016/j.cell.2020.12.004. Epub 2020 Dec 9. Cell. 2021. PMID: 33333024 Free PMC article.
Abstract
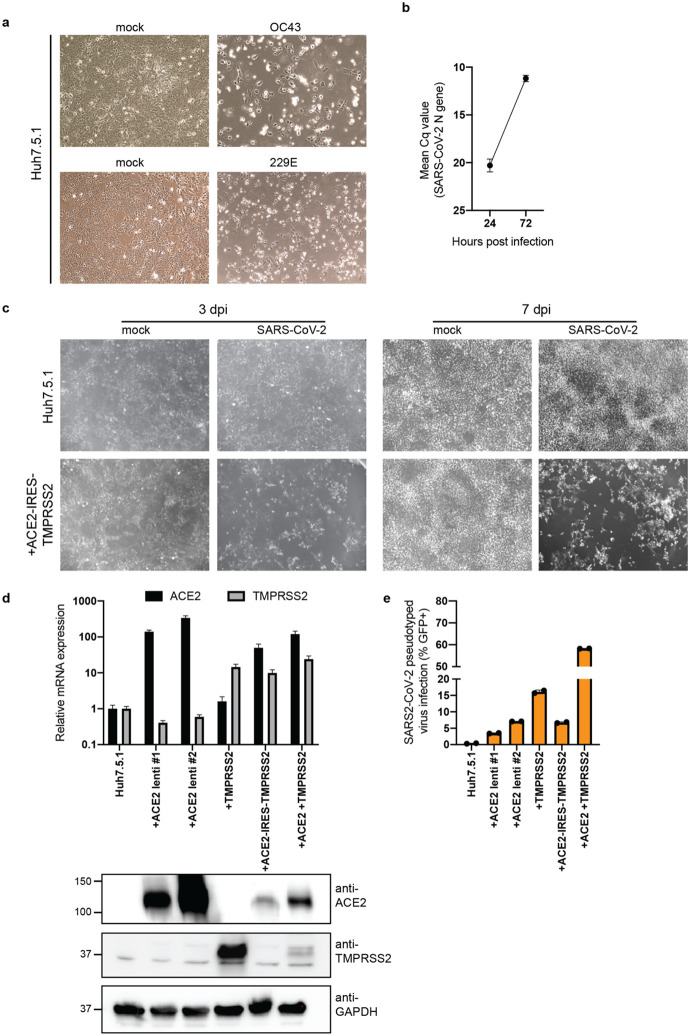
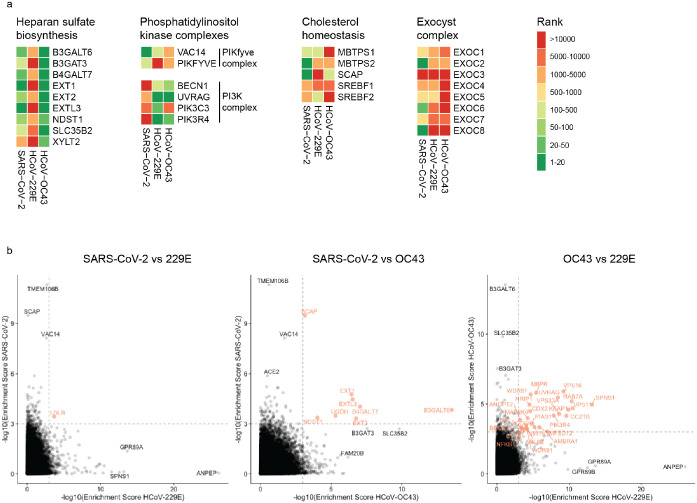
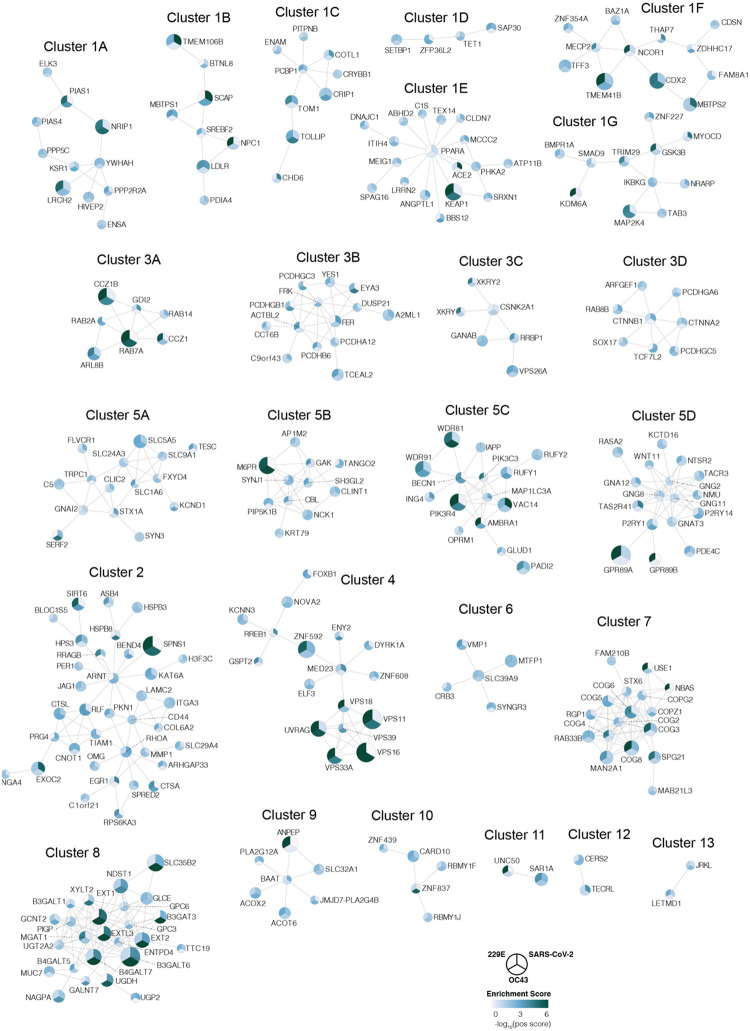
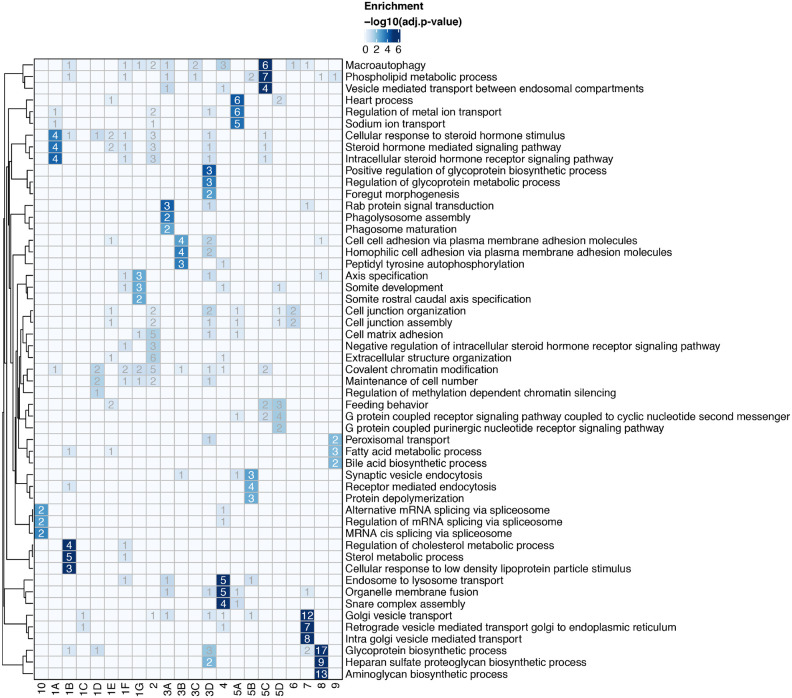
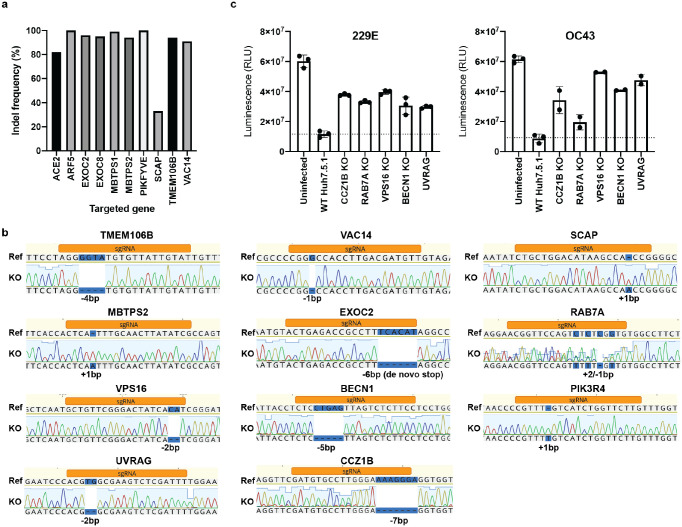
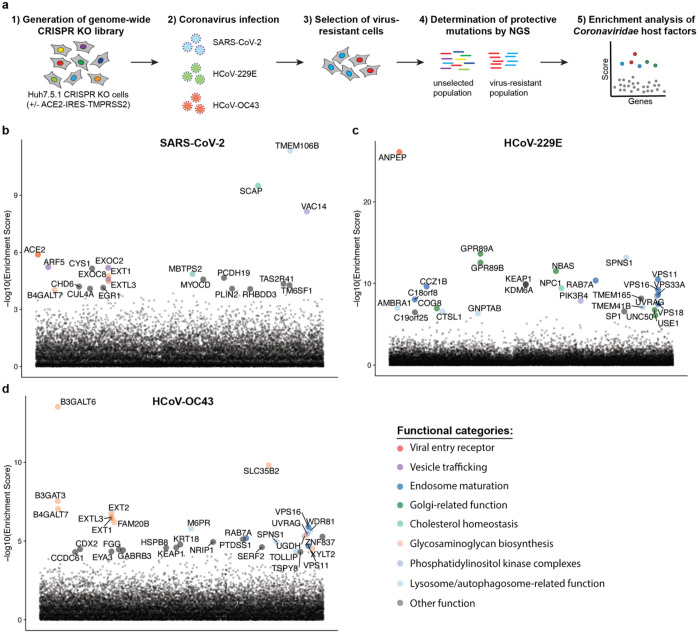
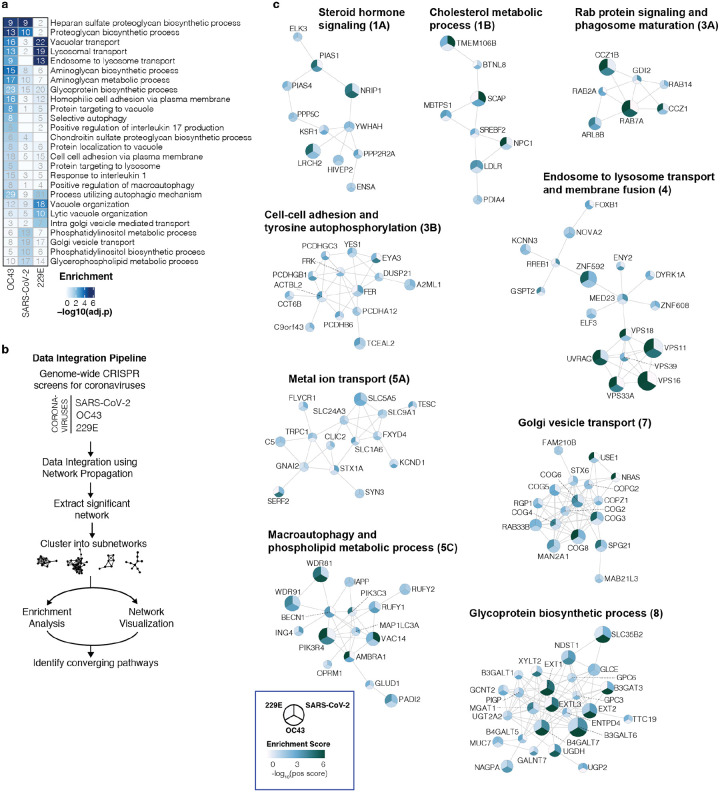
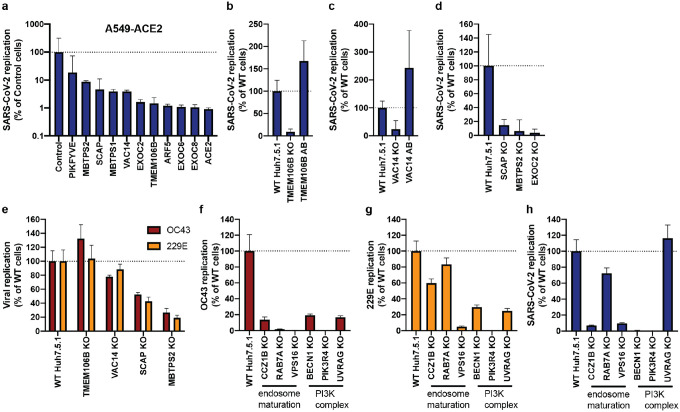
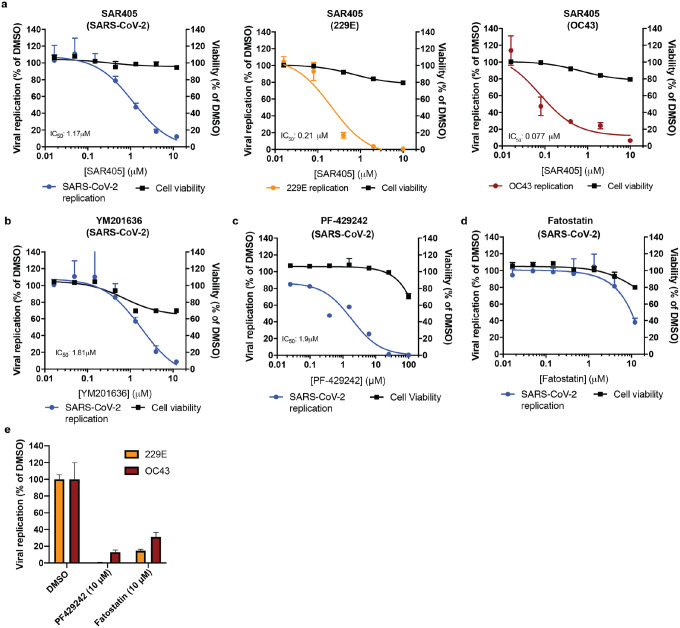
The Coronaviridae are a family of viruses that causes disease in humans ranging from mild respiratory infection to potentially lethal acute respiratory distress syndrome. Finding host factors that are common to multiple coronaviruses could facilitate the development of therapies to combat current and future coronavirus pandemics. Here, we conducted parallel genome-wide CRISPR screens in cells infected by SARS-CoV-2 as well as two seasonally circulating common cold coronaviruses, OC43 and 229E. This approach correctly identified the distinct viral entry factors ACE2 (for SARS-CoV-2), aminopeptidase N (for 229E) and glycosaminoglycans (for OC43). Additionally, we discovered phosphatidylinositol phosphate biosynthesis and cholesterol homeostasis as critical host pathways supporting infection by all three coronaviruses. By contrast, the lysosomal protein TMEM106B appeared unique to SARS-CoV-2 infection. Pharmacological inhibition of phosphatidylinositol phosphate biosynthesis and cholesterol homeostasis reduced replication of all three coronaviruses. These findings offer important insights for the understanding of the coronavirus life cycle as well as the potential development of host-directed therapies.
Conflict of interest statement
Conflict of interest
J.C.S., J.O. and K.H. are employees of Synthego Corporation.
Figures
References
-
- van der Hoek L. Human coronaviruses: what do they cause? Antivir. Ther. 12, 651–658 (2007). - PubMed
-
- Drosten C. et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 348, 1967–1976 (2003). - PubMed
-
- Zaki A. M., van Boheemen S., Bestebroer T. M., Osterhaus A. D. M. E. & Fouchier R. A. M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820 (2012). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous