

Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans
- PMID: 32997994
- PMCID: PMC7527779
- DOI: 10.1016/j.celrep.2020.108189
Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans
Abstract
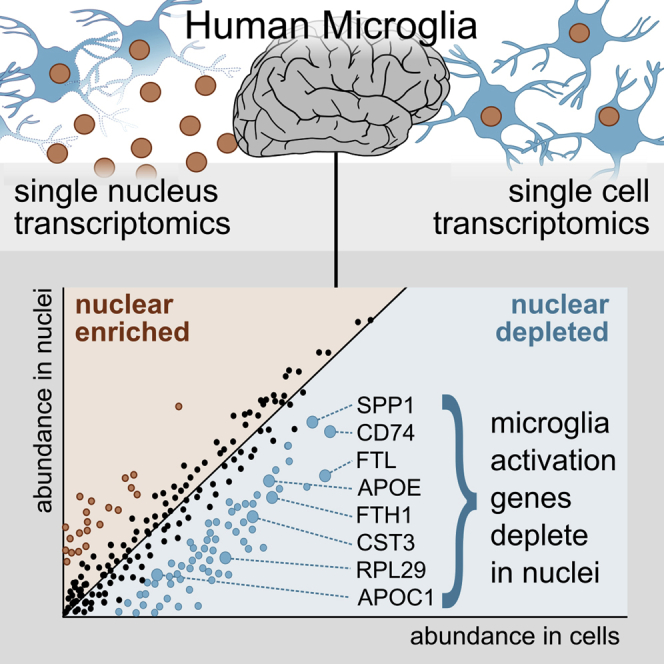
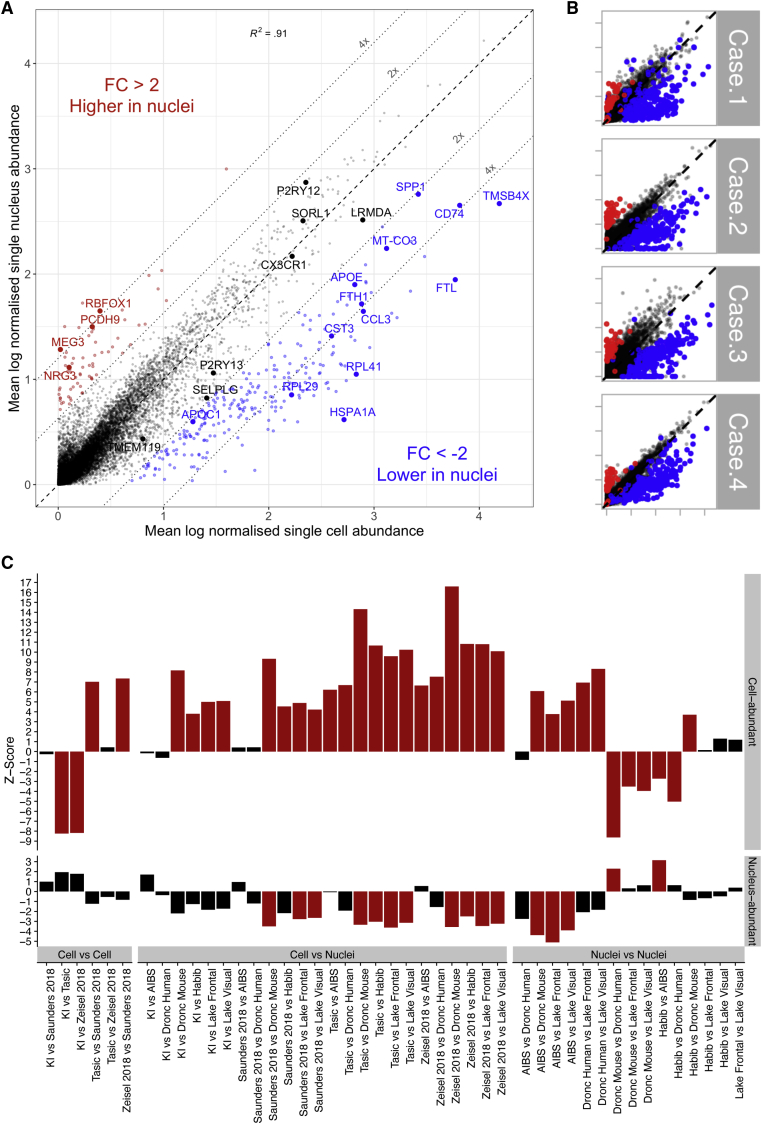
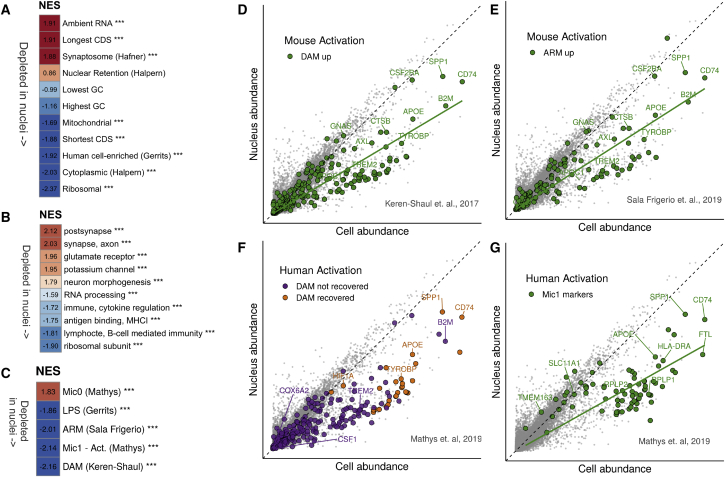
Single-nucleus RNA sequencing (snRNA-seq) is used as an alternative to single-cell RNA-seq, as it allows transcriptomic profiling of frozen tissue. However, it is unclear whether snRNA-seq is able to detect cellular state in human tissue. Indeed, snRNA-seq analyses of human brain samples have failed to detect a consistent microglial activation signature in Alzheimer's disease. Our comparison of microglia from single cells and single nuclei of four human subjects reveals that, although most genes show similar relative abundances in cells and nuclei, a small population of genes (∼1%) is depleted in nuclei compared to whole cells. This population is enriched for genes previously implicated in microglial activation, including APOE, CST3, SPP1, and CD74, comprising 18% of previously identified microglial-disease-associated genes. Given the low sensitivity of snRNA-seq to detect many activation genes, we conclude that snRNA-seq is not suited for detecting cellular activation in microglia in human disease.
Keywords: ARM; Alzheimer’s disease; activation; microglia; microglial activation; single-cell RNA-seq; single-nucleus RNA-seq.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests P.M.M. has been reimbursed for service on a Scientific Advisory Board to Ipsen Pharmaceuticals. He has received consultancy fees from Roche, Adelphi Communications, Celgene, Neurodiem, and Medscape. He has received honoraria or speakers’ fees from Novartis and Biogen and has received research or educational funds from Biogen, Novartis, and GlaxoSmithKline.
Figures
References
-
- Chen W.-T., Lu A., Craessaerts K., Pavie B., Sala Frigerio C., Corthout N., Qian X., Laláková J., Kühnemund M., Voytyuk I. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell. 2020;182:976–991.e19. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
