

The inhibitory effect of some natural bioactive compounds against SARS-CoV-2 main protease: insights from molecular docking analysis and molecular dynamic simulation
- PMID: 32998618
- PMCID: PMC7544954
- DOI: 10.1080/10934529.2020.1826192
The inhibitory effect of some natural bioactive compounds against SARS-CoV-2 main protease: insights from molecular docking analysis and molecular dynamic simulation
Abstract
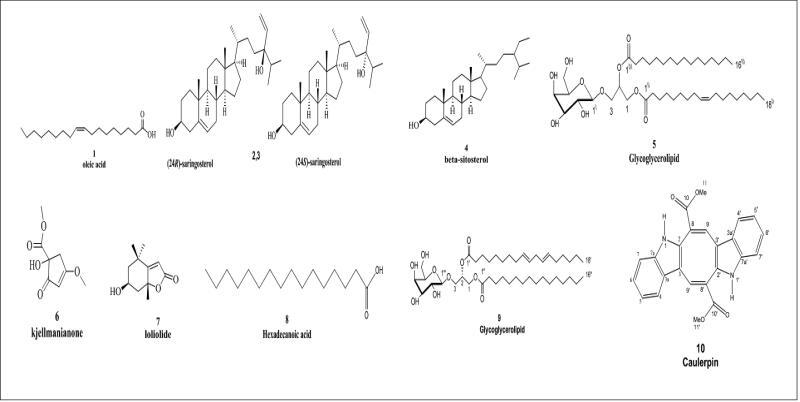
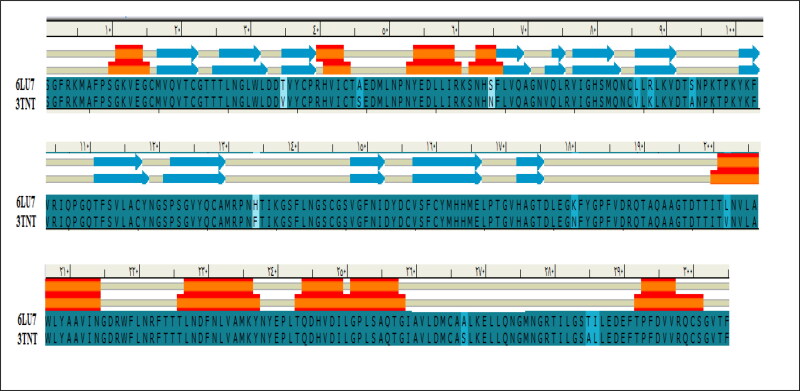
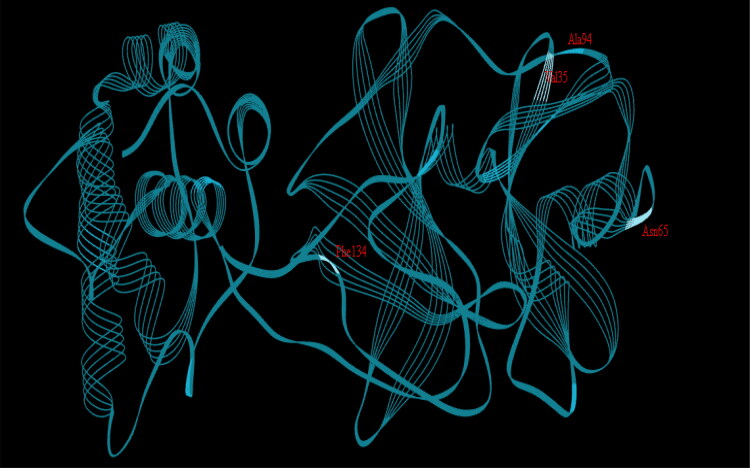
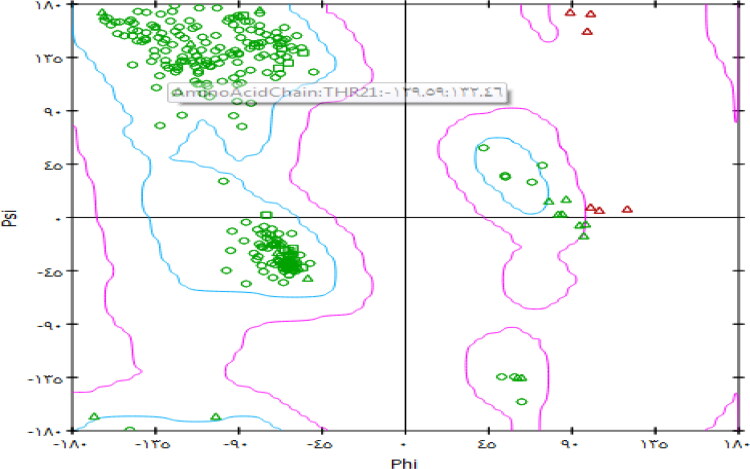
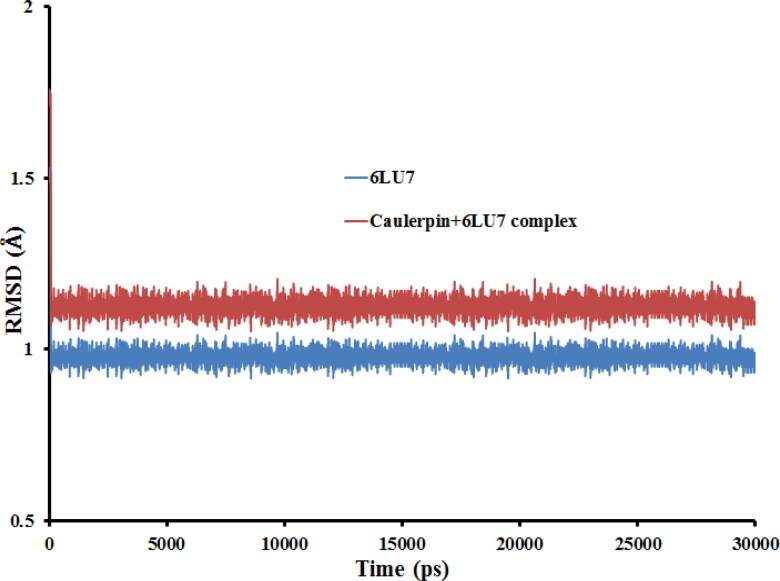
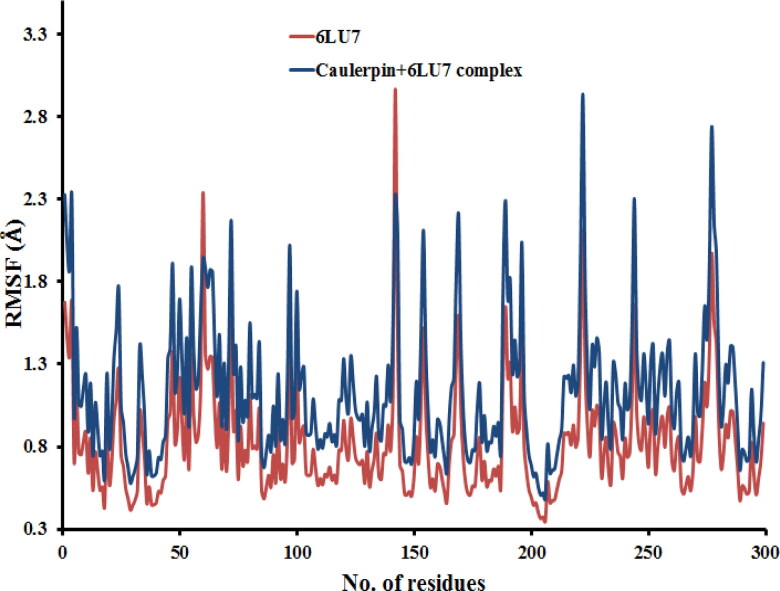
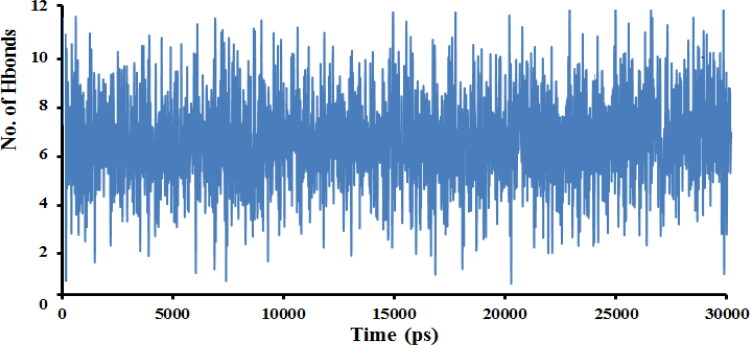
This work aimed at evaluating the inhibitory effect of ten natural bioactive compounds (1-10) as potential inhibitors of SARS-CoV-2-3CL main protease (PDB ID: 6LU7) and SARS-CoV main proteases (PDB IDs: 2GTB and 3TNT) by molecular docking analysis. The inhibitory effect of all studied compounds was studied with compared to some proposed antiviral drugs which currently used in COVID-19 treatment such as chloroquine, hydroxychloroquine, azithromycin, remdesivir, baloxvir, lopinavir, and favipiravir. Homology modeling and sequence alignment was computed to evaluate the similarity between the SARS-CoV-2-3CL main protease and other SARS-CoV receptors. ADMET properties of all studied compounds were computed and reported. Also, molecular dynamic (MD) simulation was performed on the compound which has the highest binding affinity inside 6LU7 obtained from molecular docking analysis to study it is stability inside receptor in explicit water solvent. Based on molecular docking analysis, we found that caulerpin has the highest binding affinity inside all studied receptors compared to other bioactive compounds and studied drugs. Our homology modeling and sequence alignment showed that SARS-CoV main protease (PDB ID: 3TNT) shares high similarity with 3CLpro (96.00%). Also, ADMET properties confirmed that caulerpin obeys Lipinski's rule and passes ADMET property, which make it a promising compound to act as a new safe natural drug against SARS-CoV-2-3CL main protease. Finally, MD simulation confirmed that the complex formed between caulerpin and 3CLpro is stable in water explicit and had no major effect on the flexibility of the protein throughout the simulations and provided a suitable basis for our study. Also, binding free energy between caulerpin and 6LU7 confirmed the efficacy of the caulerpin molecule against SARS-CoV-2 main protease. So, this study suggested that caulerpin could be used as a potential candidate in COVID-19 treatment.
Keywords: COVID-19 virus protease; MD simulation; caulerpin; molecular docking; natural products.
Figures



References
-
- Li, Y.; Zhang, J.; Wang, N.; Li, H.; Shi, Y.; Guo, G.; Zou, Q.. Therapeutic Drugs Targeting 2019-nCoV Main Protease by High-Throughput Screening. bioRxiv 2020.
-
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P.. Evolution of the Novel Coronavirus from the Ongoing Wuhan Outbreak and Modeling of Its Spike Protein for Risk of Human Transmission. Sci. China Life Sci. 2020, 63, 457–460. DOI: 10.1007/s11427-020-1637-5. - DOI - PMC - PubMed
-
- Lu, I.-L.; Mahindroo, N.; Liang, P.-H.; Peng, Y.-H.; Kuo, C.-J.; Tsai, K.-C.; Hsieh, H.-P.; Chao, Y.-S.; Wu, S.-Y.. Structure-Based Drug Design and Structural Biology Study of Novel Nonpeptide Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Main Protease. J. Med. Chem. 2006, 49, 5154–5161. DOI: 10.1021/jm060207o. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous