

Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy
- PMID: 32999071
- PMCID: PMC7568322
- DOI: 10.1073/pnas.2007622117
Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy
Abstract
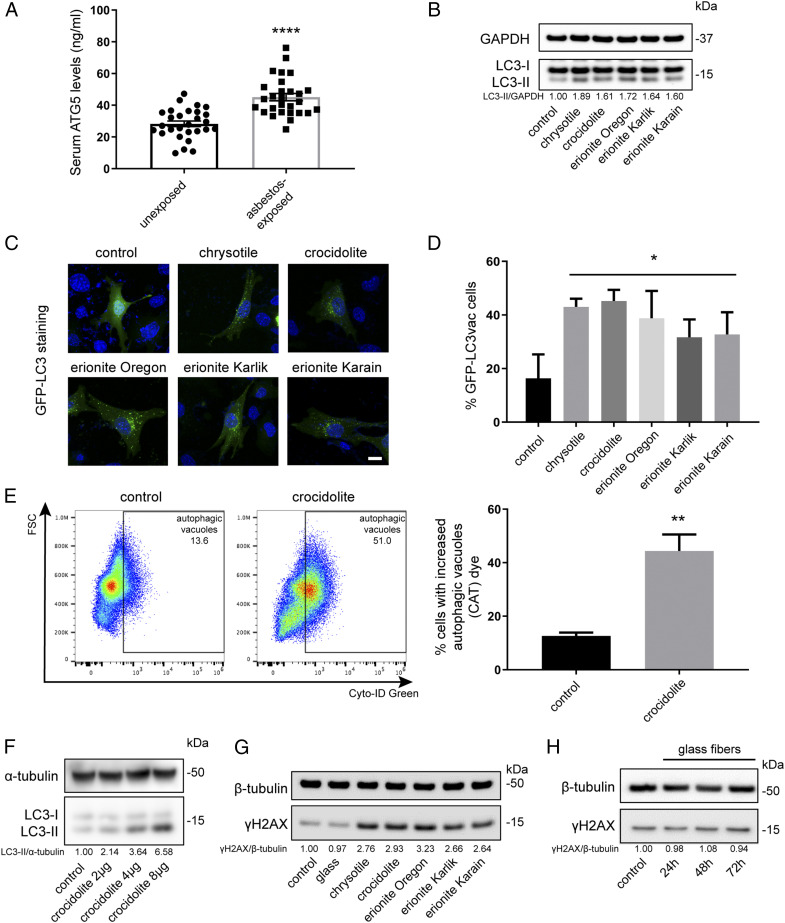
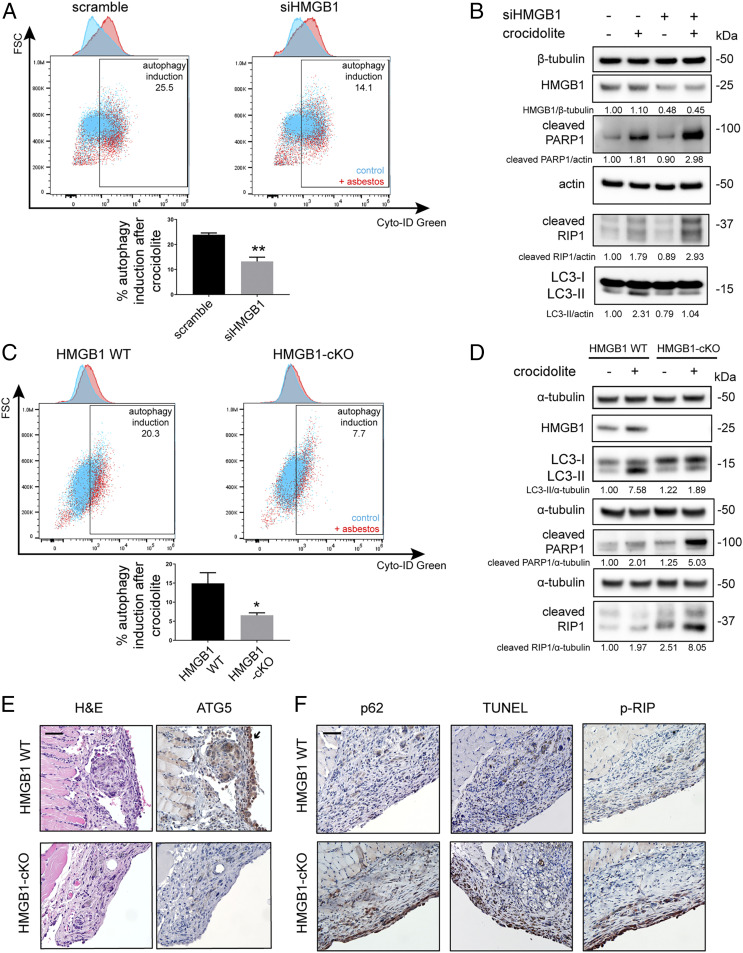
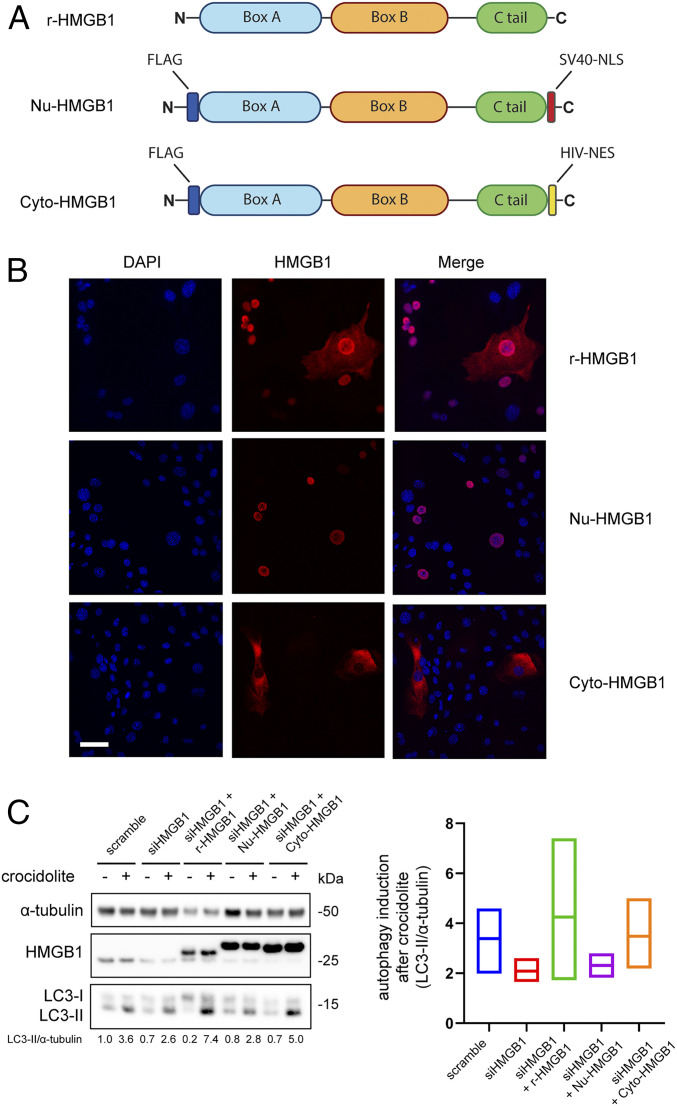
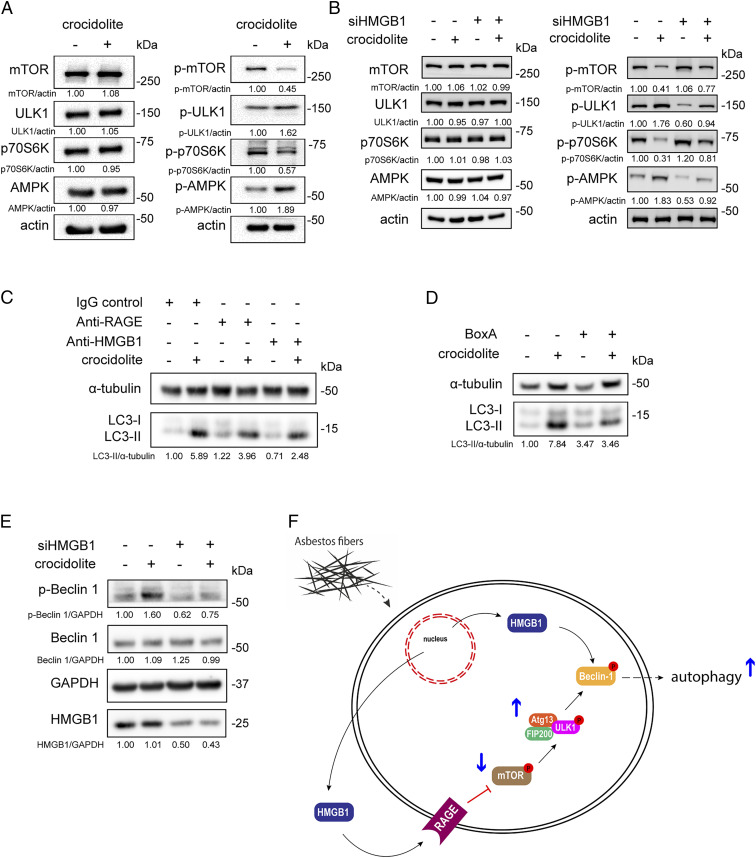
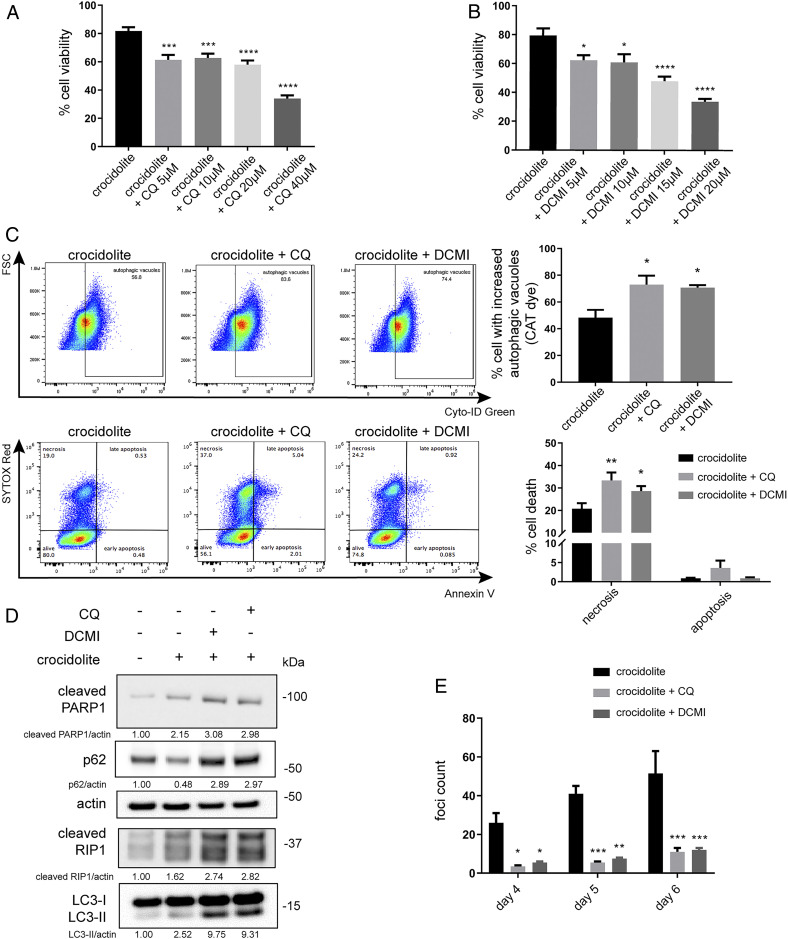
Asbestos causes malignant transformation of primary human mesothelial cells (HM), leading to mesothelioma. The mechanisms of asbestos carcinogenesis remain enigmatic, as exposure to asbestos induces HM death. However, some asbestos-exposed HM escape cell death, accumulate DNA damage, and may become transformed. We previously demonstrated that, upon asbestos exposure, HM and reactive macrophages releases the high mobility group box 1 (HMGB1) protein that becomes detectable in the tissues near asbestos deposits where HMGB1 triggers chronic inflammation. HMGB1 is also detectable in the sera of asbestos-exposed individuals and mice. Searching for additional biomarkers, we found higher levels of the autophagy marker ATG5 in sera from asbestos-exposed individuals compared to unexposed controls. As we investigated the mechanisms underlying this finding, we discovered that the release of HMGB1 upon asbestos exposure promoted autophagy, allowing a higher fraction of HM to survive asbestos exposure. HMGB1 silencing inhibited autophagy and increased asbestos-induced HM death, thereby decreasing asbestos-induced HM transformation. We demonstrate that autophagy was induced by the cytoplasmic and extracellular fractions of HMGB1 via the engagement of the RAGE receptor and Beclin 1 pathway, while nuclear HMGB1 did not participate in this process. We validated our findings in a novel unique mesothelial conditional HMGB1-knockout (HMGB1-cKO) mouse model. Compared to HMGB1 wild-type mice, mesothelial cells from HMGB1-cKO mice showed significantly reduced autophagy and increased cell death. Autophagy inhibitors chloroquine and desmethylclomipramine increased cell death and reduced asbestos-driven foci formation. In summary, HMGB1 released upon asbestos exposure induces autophagy, promoting HM survival and malignant transformation.
Keywords: HMGB1; asbestos; autophagy; cell death; mesothelioma.
Conflict of interest statement
Competing interest statement: M.C. has a patent issued for BAP1. M.C. and H.Y. have two patents issued for HMGB1. M.C. is a board-certified pathologist who provides consultation for pleural pathology, including medical–legal consultation.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
