

The bacterial sulfur cycle in expanding dysoxic and euxinic marine waters
- PMID: 33000514
- PMCID: PMC8359478
- DOI: 10.1111/1462-2920.15265
The bacterial sulfur cycle in expanding dysoxic and euxinic marine waters
Abstract
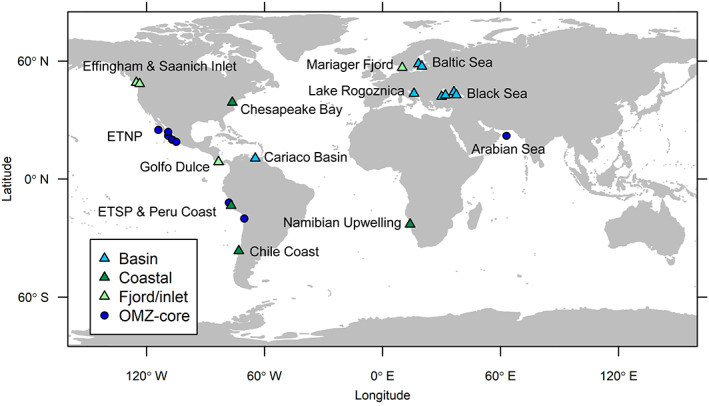
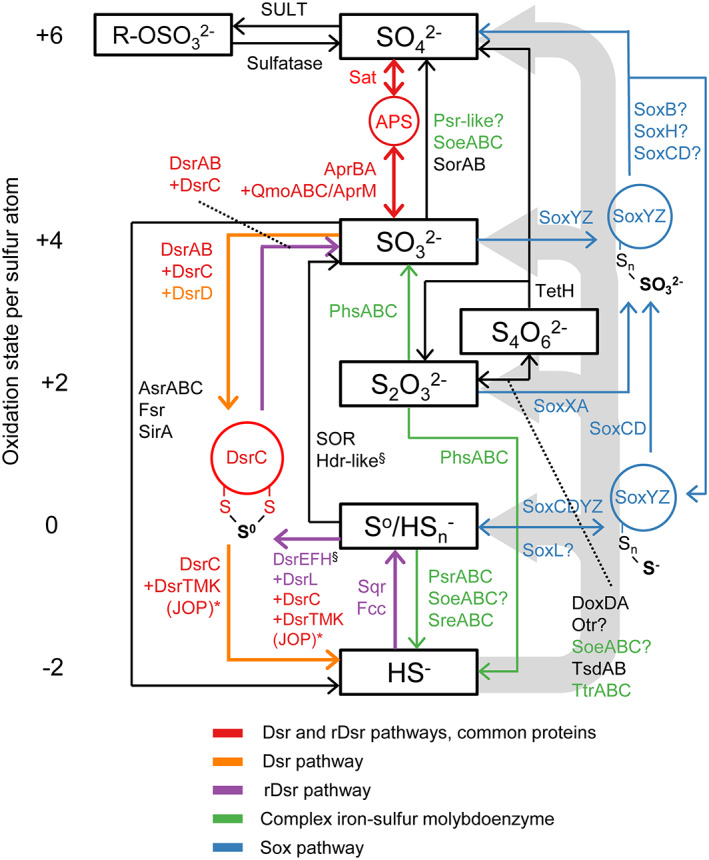
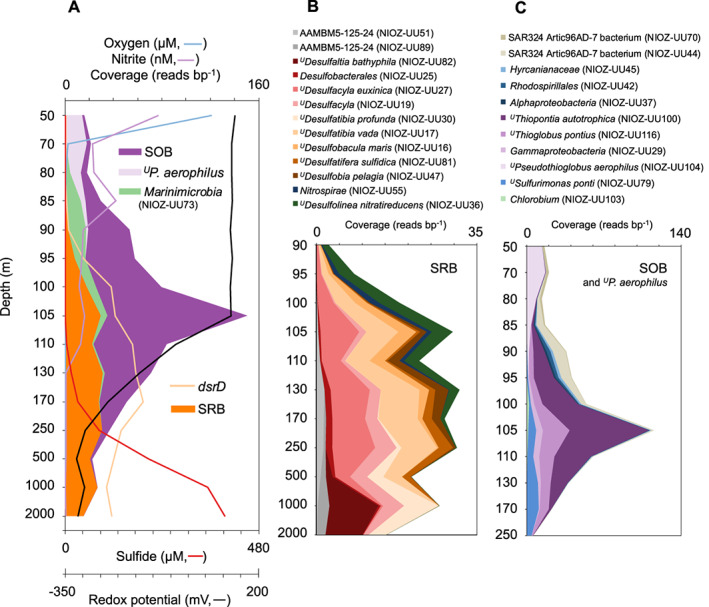
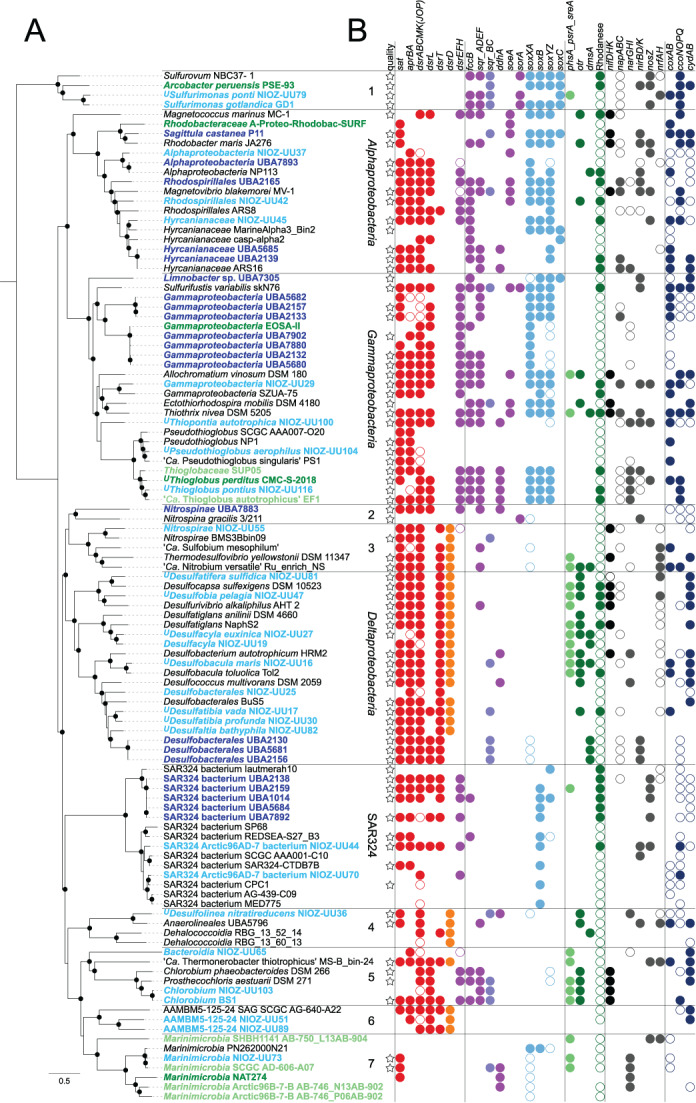
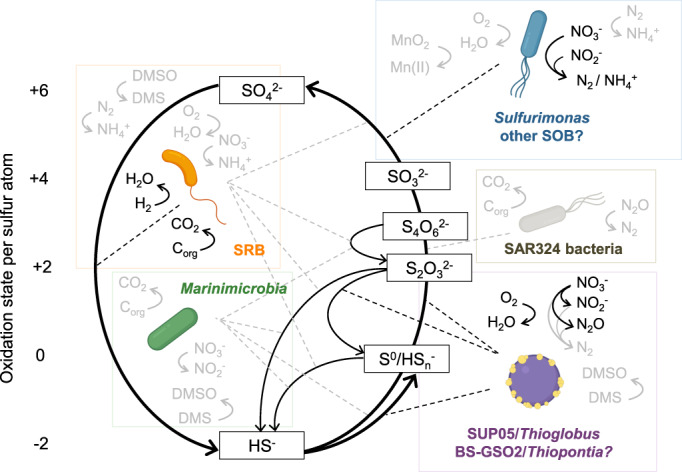
Dysoxic marine waters (DMW, < 1 μM oxygen) are currently expanding in volume in the oceans, which has biogeochemical, ecological and societal consequences on a global scale. In these environments, distinct bacteria drive an active sulfur cycle, which has only recently been recognized for open-ocean DMW. This review summarizes the current knowledge on these sulfur-cycling bacteria. Critical bottlenecks and questions for future research are specifically addressed. Sulfate-reducing bacteria (SRB) are core members of DMW. However, their roles are not entirely clear, and they remain largely uncultured. We found support for their remarkable diversity and taxonomic novelty by mining metagenome-assembled genomes from the Black Sea as model ecosystem. We highlight recent insights into the metabolism of key sulfur-oxidizing SUP05 and Sulfurimonas bacteria, and discuss the probable involvement of uncultivated SAR324 and BS-GSO2 bacteria in sulfur oxidation. Uncultivated Marinimicrobia bacteria with a presumed organoheterotrophic metabolism are abundant in DMW. Like SRB, they may use specific molybdoenzymes to conserve energy from the oxidation, reduction or disproportionation of sulfur cycle intermediates such as S0 and thiosulfate, produced from the oxidation of sulfide. We expect that tailored sampling methods and a renewed focus on cultivation will yield deeper insight into sulfur-cycling bacteria in DMW.
© 2020 The Authors. Environmental Microbiology published by Society for Applied Microbiology and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Genomic evidence for sulfur intermediates as new biogeochemical hubs in a model aquatic microbial ecosystem.Microbiome. 2021 Feb 16;9(1):46. doi: 10.1186/s40168-021-00999-x. Microbiome. 2021. PMID: 33593438 Free PMC article.
-
A cryptic sulfur cycle in oxygen-minimum-zone waters off the Chilean coast.Science. 2010 Dec 3;330(6009):1375-8. doi: 10.1126/science.1196889. Epub 2010 Nov 11. Science. 2010. PMID: 21071631
-
Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones.Science. 2009 Oct 23;326(5952):578-82. doi: 10.1126/science.1175309. Science. 2009. PMID: 19900896
-
Sulfur-oxidizing bacteria (SOB) and sulfate-reducing bacteria (SRB) in oil reservoir and biological control of SRB: a review.Arch Microbiol. 2023 Apr 3;205(5):162. doi: 10.1007/s00203-023-03520-0. Arch Microbiol. 2023. PMID: 37010699 Review.
-
The Biogeochemical Sulfur Cycle of Marine Sediments.Front Microbiol. 2019 Apr 24;10:849. doi: 10.3389/fmicb.2019.00849. eCollection 2019. Front Microbiol. 2019. PMID: 31105660 Free PMC article. Review.
Cited by
-
Global diversity and inferred ecophysiology of microorganisms with the potential for dissimilatory sulfate/sulfite reduction.FEMS Microbiol Rev. 2023 Sep 5;47(5):fuad058. doi: 10.1093/femsre/fuad058. FEMS Microbiol Rev. 2023. PMID: 37796897 Free PMC article.
-
Generation and Physiology of Hydrogen Sulfide and Reactive Sulfur Species in Bacteria.Antioxidants (Basel). 2022 Dec 17;11(12):2487. doi: 10.3390/antiox11122487. Antioxidants (Basel). 2022. PMID: 36552695 Free PMC article. Review.
-
Redox gradient shapes the abundance and diversity of mercury-methylating microorganisms along the water column of the Black Sea.mSystems. 2023 Aug 31;8(4):e0053723. doi: 10.1128/msystems.00537-23. Epub 2023 Aug 14. mSystems. 2023. PMID: 37578240 Free PMC article.
-
The 'oxygen' in oxygen minimum zones.Environ Microbiol. 2022 Nov;24(11):5332-5344. doi: 10.1111/1462-2920.16192. Epub 2022 Oct 18. Environ Microbiol. 2022. PMID: 36054074 Free PMC article. Review.
-
Study of Microbial Sulfur Metabolism in a Near Real-Time Pathway through Confocal Raman Quantitative 3D Imaging.Microbiol Spectr. 2023 Feb 21;11(2):e0367822. doi: 10.1128/spectrum.03678-22. Online ahead of print. Microbiol Spectr. 2023. PMID: 36809047 Free PMC article.
References
-
- Albert, D.B., Taylor, C., and Martens, C.S. (1995) Sulfate reduction rates and low‐molecular‐weight fatty‐acid concentrations in the water column and surficial sediments of the Black Sea. Deep Sea Res Part I Oceanogr Res Pap 42: 1239–1260.
-
- Alldredge, A.L., and Silver, M.W. (1988) Characteristics, dynamics and significance of marine snow. Prog Oceanogr 20: 41–82.
-
- Anantharaman, K., Duhaime, M.B., Breier, J.A., Wendt, K.A., Toner, B.M., and Dick, G.J. (2014) Sulfur oxidation genes in diverse deep‐sea viruses. Science 344: 757–760. - PubMed
-
- Anderson, R., Wylezich, C., Glaubitz, S., Labrenz, M., and Jurgens, K. (2013) Impact of protist grazing on a key bacterial group for biogeochemical cycling in Baltic Sea pelagic oxic/anoxic interfaces. Environ Microbiol 15: 1580–1594. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
