

Barth syndrome cardiomyopathy: targeting the mitochondria with elamipretide
- PMID: 33001359
- PMCID: PMC7895793
- DOI: 10.1007/s10741-020-10031-3
Barth syndrome cardiomyopathy: targeting the mitochondria with elamipretide
Abstract
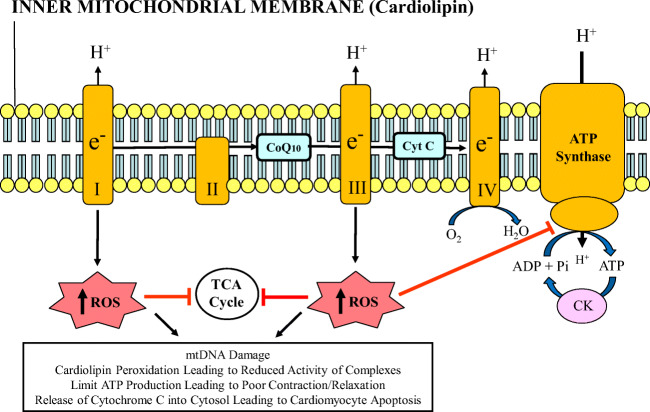
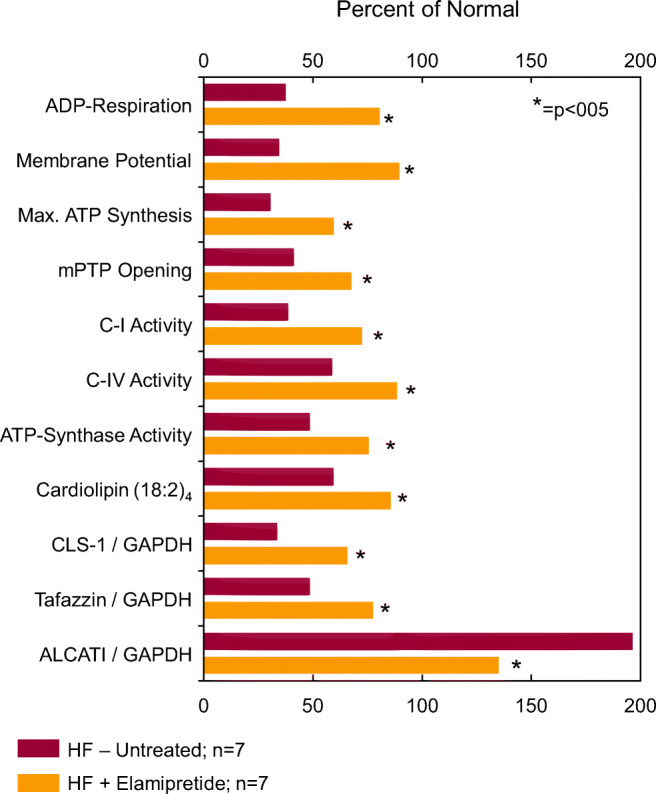
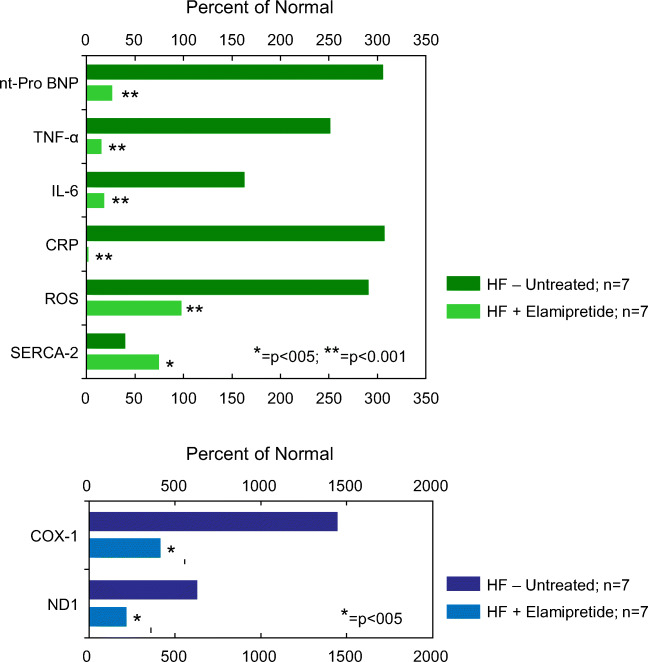
Barth syndrome (BTHS) is a rare, X-linked recessive, infantile-onset debilitating disorder characterized by early-onset cardiomyopathy, skeletal muscle myopathy, growth delay, and neutropenia, with a worldwide incidence of 1/300,000-400,000 live births. The high mortality rate throughout infancy in BTHS patients is related primarily to progressive cardiomyopathy and a weakened immune system. BTHS is caused by defects in the TAZ gene that encodes tafazzin, a transacylase responsible for the remodeling and maturation of the mitochondrial phospholipid cardiolipin (CL), which is critical to normal mitochondrial structure and function (i.e., ATP generation). A deficiency in tafazzin results in up to a 95% reduction in levels of structurally mature CL. Because the heart is the most metabolically active organ in the body, with the highest mitochondrial content of any tissue, mitochondrial dysfunction plays a key role in the development of heart failure in patients with BTHS. Changes in mitochondrial oxidative phosphorylation reduce the ability of mitochondria to meet the ATP demands of the human heart as well as skeletal muscle, namely ATP synthesis does not match the rate of ATP consumption. The presence of several cardiomyopathic phenotypes have been described in BTHS, including dilated cardiomyopathy, left ventricular noncompaction, either alone or in conjunction with other cardiomyopathic phenotypes, endocardial fibroelastosis, hypertrophic cardiomyopathy, and an apical form of hypertrophic cardiomyopathy, among others, all of which can be directly attributed to the lack of CL synthesis, remodeling, and maturation with subsequent mitochondrial dysfunction. Several mechanisms by which these cardiomyopathic phenotypes exist have been proposed, thereby identifying potential targets for treatment. Dysfunction of the sarcoplasmic reticulum Ca2+-ATPase pump and inflammation potentially triggered by circulating mitochondrial components have been identified. Currently, treatment modalities are aimed at addressing symptomatology of HF in BTHS, but do not address the underlying pathology. One novel therapeutic approach includes elamipretide, which crosses the mitochondrial outer membrane to localize to the inner membrane where it associates with cardiolipin to enhance ATP synthesis in several organs, including the heart. Encouraging clinical results of the use of elamipretide in treating patients with BTHS support the potential use of this drug for management of this rare disease.
Keywords: Adenosine triphosphate; Barth syndrome; Cardiomyopathies; Electron transport chain; Mitochondria.
Figures
References
-
- Dubek J, Maack C. Barth syndrome cardiomyopathy. Cardiovasc Res. 2017;113:399–410. - PubMed
-
- Barth P, Van den Bogert C, Bolhuis P, Scholte H, van Gennip A, Schutgens R, Ketel A. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): respiratory-chain abnormalities in cultured fibroblasts. J Inherit Metab Dis. 1996;19:157–160. - PubMed
-
- Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. 1996;12:385–389. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
