

Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing
- PMID: 33001864
- PMCID: PMC7773386
- DOI: 10.1172/JCI141500
Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing
Abstract
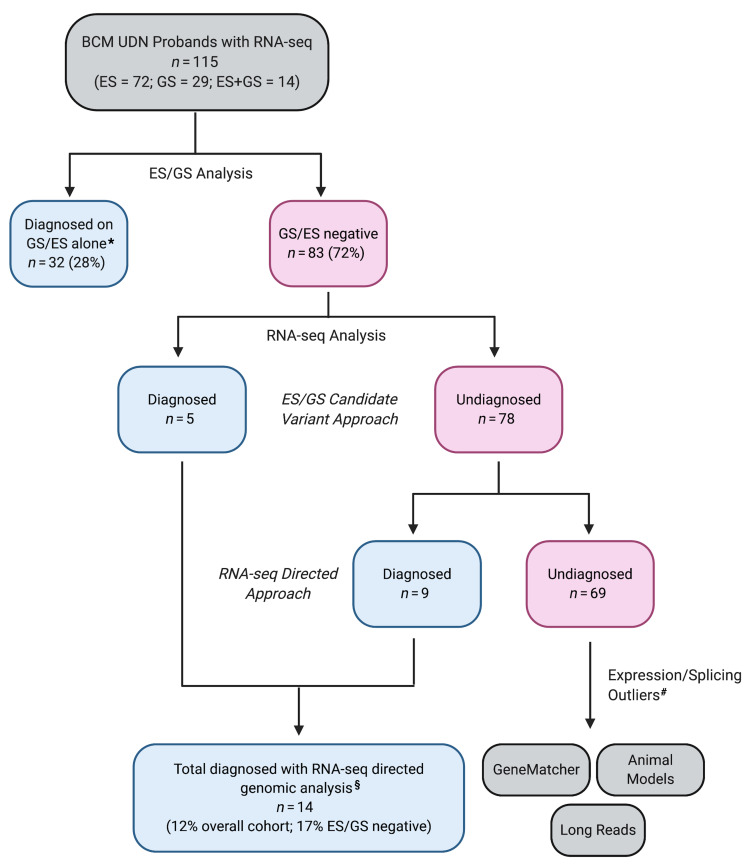
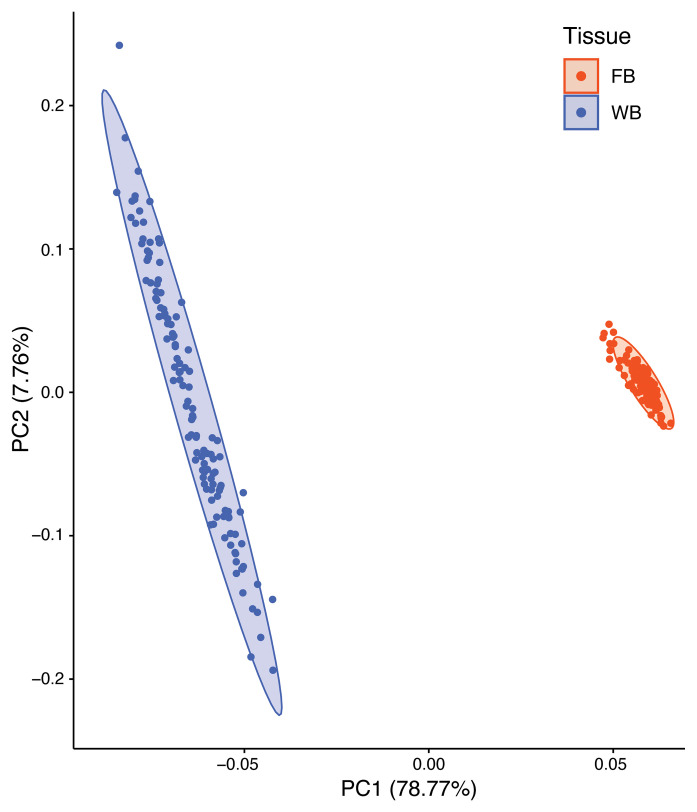
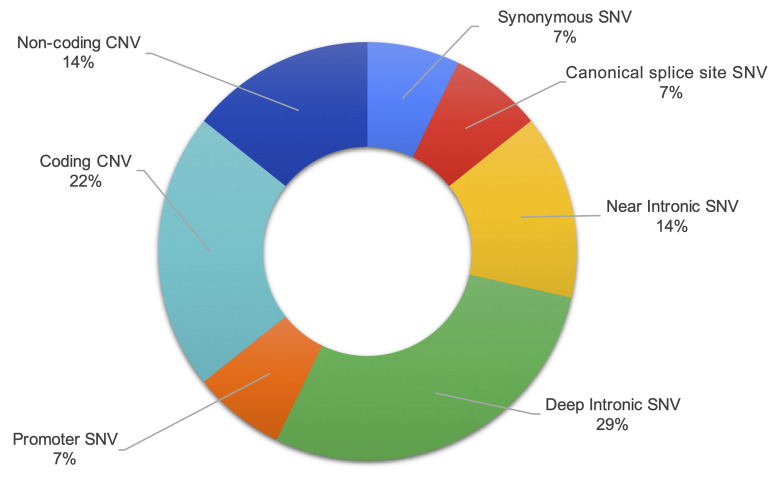
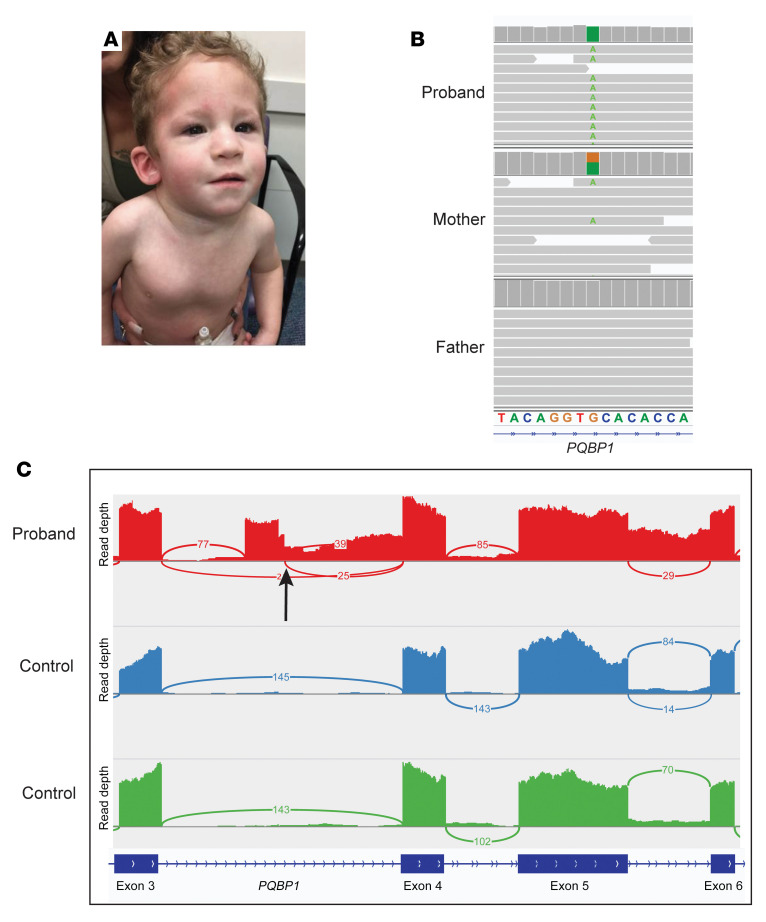
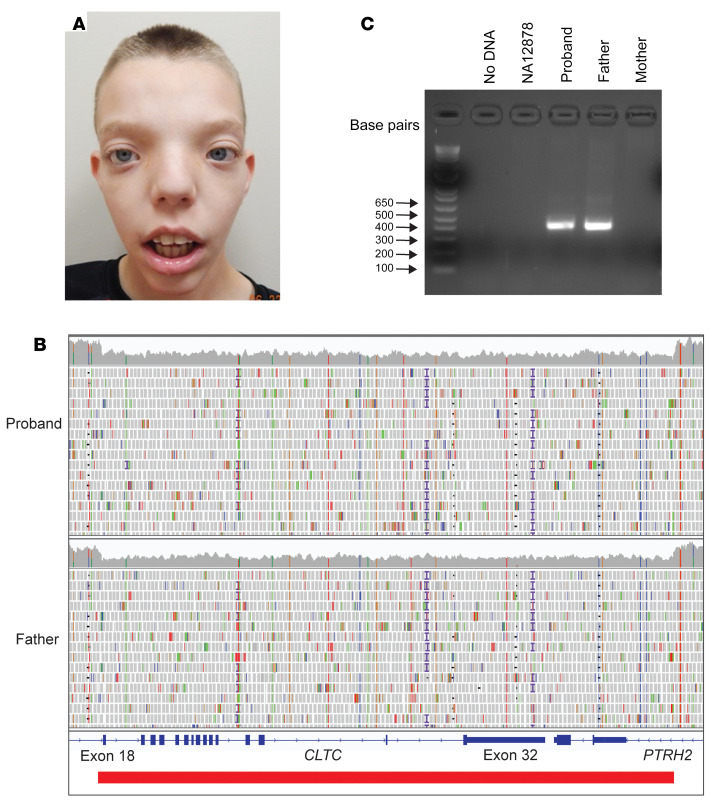
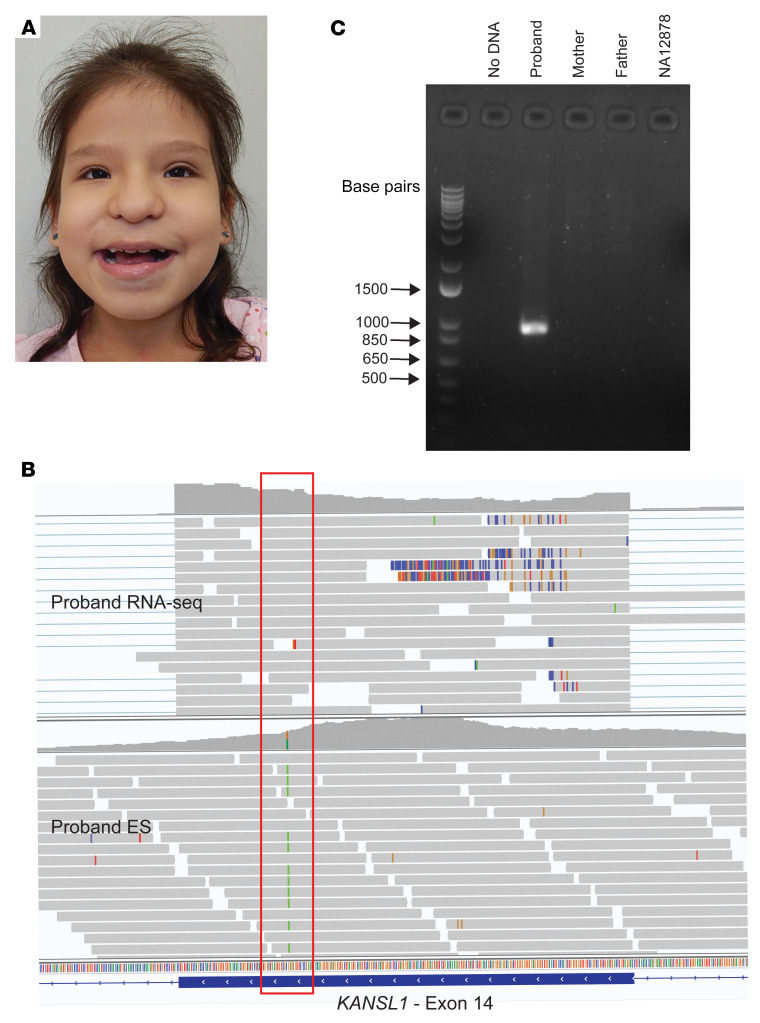
BACKGROUNDTranscriptome sequencing (RNA-seq) improves diagnostic rates in individuals with suspected Mendelian conditions to varying degrees, primarily by directing the prioritization of candidate DNA variants identified on exome or genome sequencing (ES/GS). Here we implemented an RNA-seq-guided method to diagnose individuals across a wide range of ages and clinical phenotypes.METHODSOne hundred fifteen undiagnosed adult and pediatric patients with diverse phenotypes and 67 family members (182 total individuals) underwent RNA-seq from whole blood and skin fibroblasts at the Baylor College of Medicine (BCM) Undiagnosed Diseases Network clinical site from 2014 to 2020. We implemented a workflow to detect outliers in gene expression and splicing for cases that remained undiagnosed despite standard genomic and transcriptomic analysis.RESULTSThe transcriptome-directed approach resulted in a diagnostic rate of 12% across the entire cohort, or 17% after excluding cases solved on ES/GS alone. Newly diagnosed conditions included Koolen-de Vries syndrome (KANSL1), Renpenning syndrome (PQBP1), TBCK-associated encephalopathy, NSD2- and CLTC-related intellectual disability, and others, all with negative conventional genomic testing, including ES and chromosomal microarray (CMA). Skin fibroblasts exhibited higher and more consistent expression of clinically relevant genes than whole blood. In solved cases with RNA-seq from both tissues, the causative defect was missed in blood in half the cases but none from fibroblasts.CONCLUSIONSFor our cohort of undiagnosed individuals with suspected Mendelian conditions, transcriptome-directed genomic analysis facilitated diagnoses, primarily through the identification of variants missed on ES and CMA.TRIAL REGISTRATIONNot applicable.FUNDINGNIH Common Fund, BCM Intellectual and Developmental Disabilities Research Center, Eunice Kennedy Shriver National Institute of Child Health & Human Development.
Keywords: Diagnostics; Genetic diseases; Genetics; Molecular genetics.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
