

Novel Approaches to Target Mutant FLT3 Leukaemia
- PMID: 33003568
- PMCID: PMC7600363
- DOI: 10.3390/cancers12102806
Novel Approaches to Target Mutant FLT3 Leukaemia
Abstract
Fms-like tyrosine kinase 3 (FLT3) is a member of the class III receptor tyrosine kinases (RTK) and is involved in cell survival, proliferation, and differentiation of haematopoietic progenitors of lymphoid and myeloid lineages. Oncogenic mutations in the FLT3 gene resulting in constitutively active FLT3 variants are frequently found in acute myeloid leukaemia (AML) patients and correlate with patient's poor survival. Targeting FLT3 mutant leukaemic stem cells (LSC) is a key to efficient treatment of patients with relapsed/refractory AML. It is therefore essential to understand how LSC escape current therapies in order to develop novel therapeutic strategies. Here, we summarize the current knowledge on mechanisms of FLT3 activity regulation and its cellular consequences. Furthermore, we discuss how aberrant FLT3 signalling cooperates with other oncogenic lesions and the microenvironment to drive haematopoietic malignancies and how this can be harnessed for therapeutical purposes.
Keywords: FMS-like tyrosine kinase 3 (FLT3); acute myeloid leukaemia (AML); cancer cell vulnerability; haematopoietic niche; oncogenic signaling; re-sistance development.
Conflict of interest statement
The other authors declare that they have no conflict of interest.
Figures
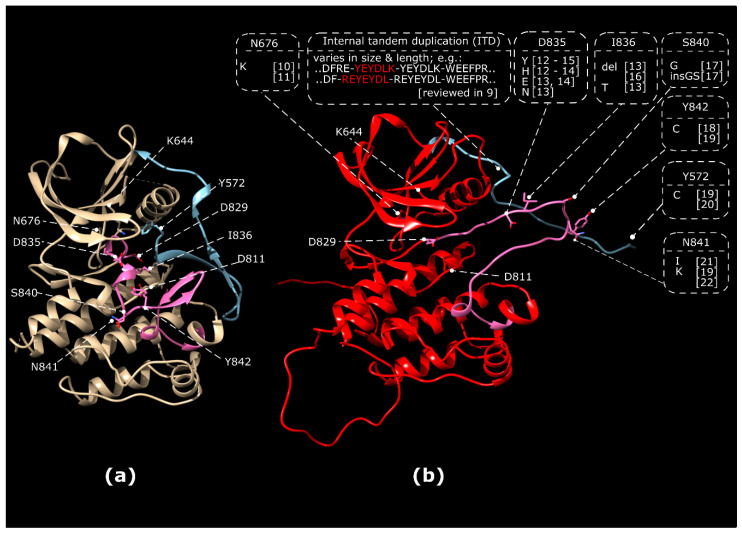
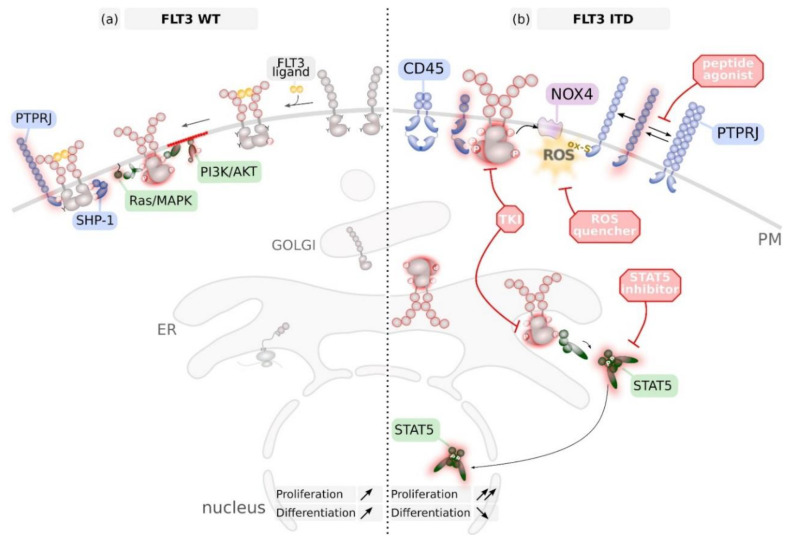
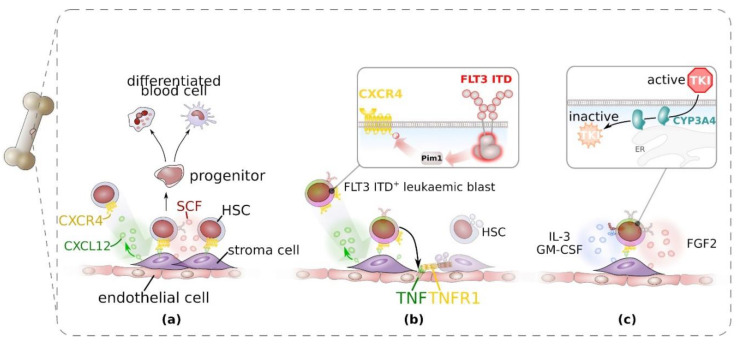
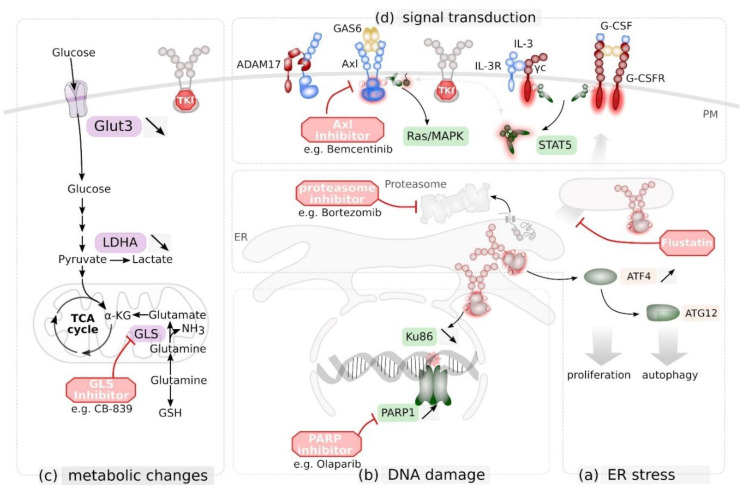
References
-
- Maroc N., Rottapel R., Rosnet O., Marchetto S., Lavezzi C., Mannoni P., Birnbaum D., Dubreuil P. Biochemical characterization and analysis of the transforming potential of the FLT3/FLK2 receptor tyrosine kinase. Oncogene. 1993;8:909–918. - PubMed
-
- Kikushige Y., Yoshimoto G., Miyamoto T., Iino T., Mori Y., Iwasaki H., Niiro H., Takenaka K., Nagafuji K., Harada M., et al. Human Flt3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J. Immunol. 2008;180:7358–7367. doi: 10.4049/jimmunol.180.11.7358. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
