

The allosteric activation mechanism of a phospholipase A2-like toxin from Bothrops jararacussu venom: a dynamic description
- PMID: 33004851
- PMCID: PMC7529814
- DOI: 10.1038/s41598-020-73134-9
The allosteric activation mechanism of a phospholipase A2-like toxin from Bothrops jararacussu venom: a dynamic description
Abstract
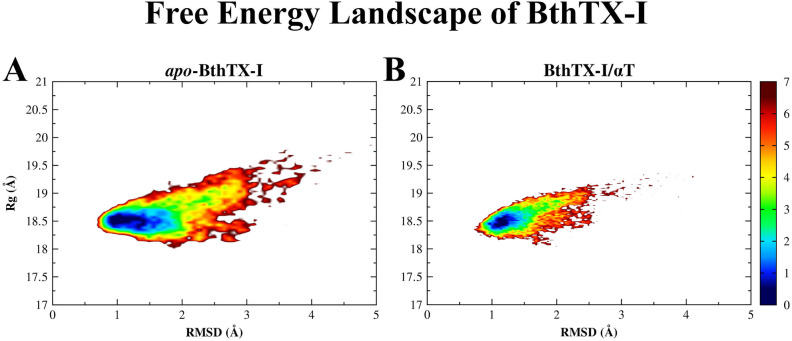
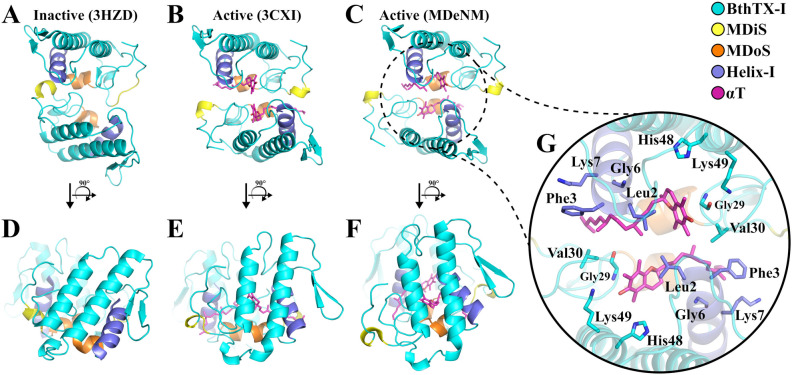
The activation process of phospholipase A2-like (PLA2-like) toxins is a key step in their molecular mechanism, which involves oligomeric changes leading to the exposure of specific sites. Few studies have focused on the characterization of allosteric activators and the features that distinguish them from inhibitors. Herein, a comprehensive study with the BthTX-I toxin from Bothrops jararacussu venom bound or unbound to α-tocopherol (αT) was carried out. The oligomerization state of BthTX-I bound or unbound to αT in solution was studied and indicated that the toxin is predominantly monomeric but tends to oligomerize when complexed with αT. In silico molecular simulations showed the toxin presents higher conformational changes in the absence of αT, which suggests that it is important to stabilize the structure of the toxin. The transition between the two states (active/inactive) was also studied, showing that only the unbound BthTX-I system could migrate to the inactive state. In contrast, the presence of αT induces the toxin to leave the inactive state, guiding it towards the active state, with more regions exposed to the solvent, particularly its active site. Finally, the structural determinants necessary for a molecule to be an inhibitor or activator were analyzed in light of the obtained results.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- World Heath Organization, Neglected tropical diseases, https://www.who.int/neglected_diseases/en/ (2018).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
