

Primary carnitine deficiency in two sisters with intractable epilepsy and reversible metabolic cardiomyopathy: Two case reports
- PMID: 33005244
- PMCID: PMC7523286
- DOI: 10.3892/etm.2020.9246
Primary carnitine deficiency in two sisters with intractable epilepsy and reversible metabolic cardiomyopathy: Two case reports
Abstract
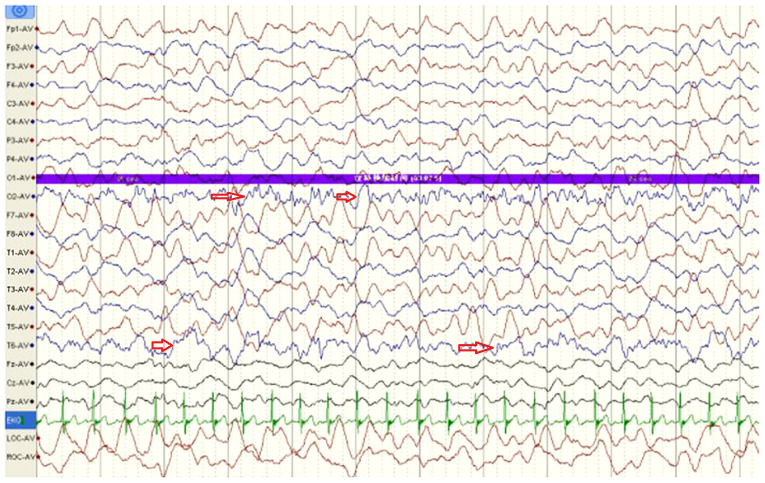
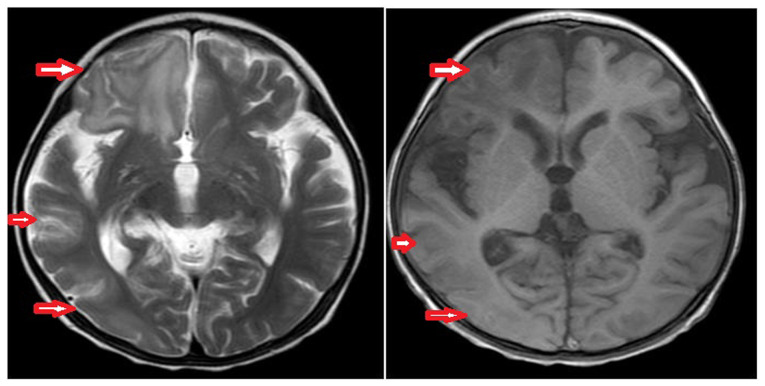
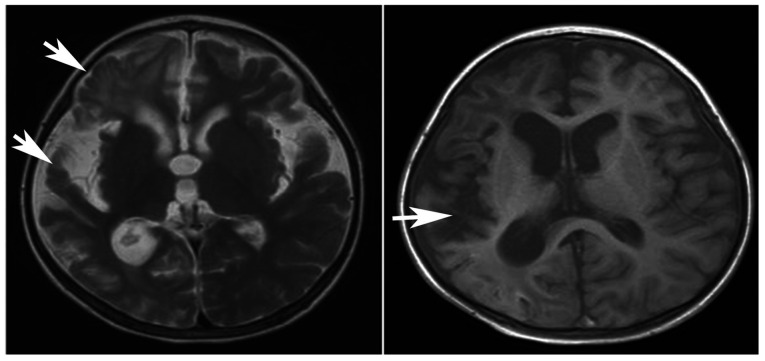
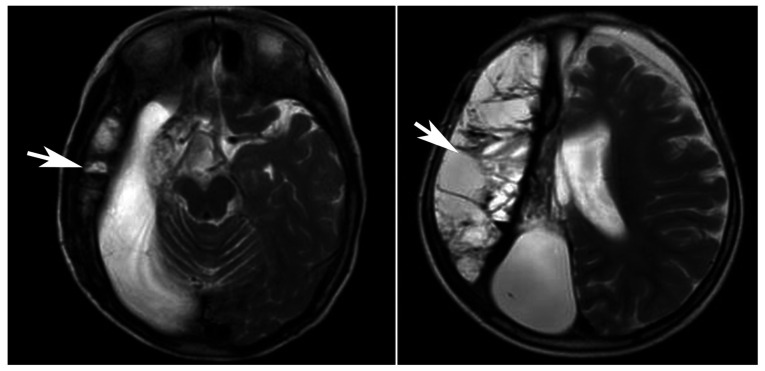
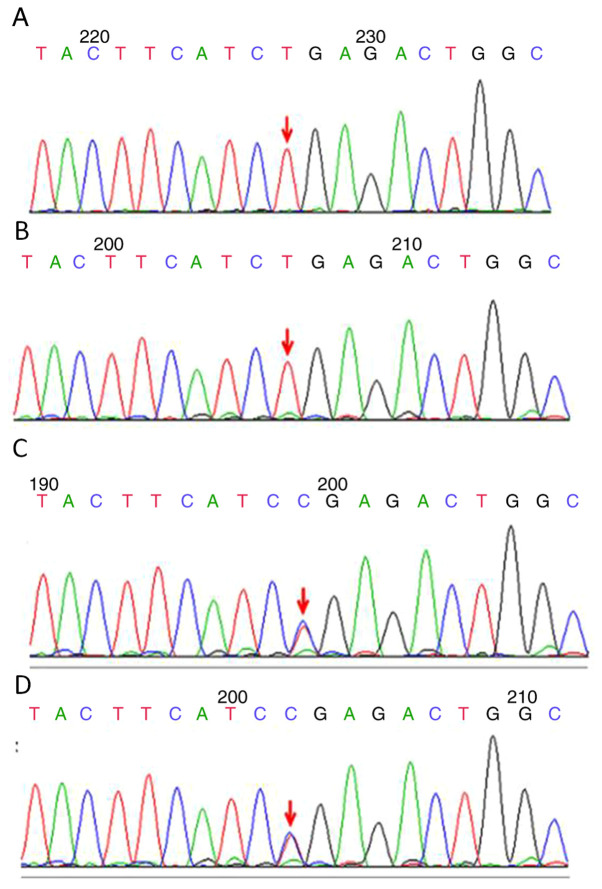
Primary carnitine deficiency (PCD) is a disorder of the carnitine cycle that results in defective fatty acid oxidation. When carnitine cannot be transported into the cells, fatty acid oxidation is impaired, resulting a variety of symptoms, such as chronic muscle weakness, cardiomyopathy, hypoglycemia and liver dysfunction. The clinical manifestations and outcomes of different cases with PCD vary among patients. The present case report focused on two sisters with PCD. The younger sister presented with intractable epilepsy, and the older sister presented with reversible metabolic cardiomyopathy. Potential mutations in the SLC22A5 gene were investigated within the family, and a nonsense mutation [c.760C>T (p.R254X)] was identified in four family members. The two sisters harbored homozygous mutations, whereas their parents presented heterozygous mutations. Metabolic disease screening revealed low plasma free carnitine levels (<5 µmol/l) in the two sisters. The plasma free carnitine levels of their parents were normal, and they were asymptomatic. PCD in the two patients was managed using oral levocarnitine. The metabolic cardiomyopathy of the older sister improved following 3 months of treatment. However, the epilepsy of the younger sister was recurrent with oral antiepileptic therapy lasting one year and eight months, and epilepsy was finally controlled following right cerebral resection. The present case report demonstrated that the clinical manifestations presented by patients with PCD within the same family were different. The results indicated that treatment with levocarnitine supplementation should be initiated as soon as possible before irreversible organ damage occurs. In addition, metabolic decompensation and cardiac muscle functions were improved following carnitine supplementation. The resection of the severely diseased unilateral brain combined with carnitine supplementation and antiepileptic therapy may be an effective treatment for PCD with intractable epilepsy complications.
Keywords: PCD; cardiomyopathy; encephalopathy; gene mutation.
Copyright: © Yang et al.
Figures
Similar articles
-
A report of a pedigree with compound heterozygous mutations in the SLC22A5 gene.Front Pediatr. 2023 Jun 6;11:985720. doi: 10.3389/fped.2023.985720. eCollection 2023. Front Pediatr. 2023. PMID: 37351314 Free PMC article.
-
Infantile primary carnitine deficiency: A severe cardiac presentation unresponsive to carnitine supplementation.JIMD Rep. 2022 Nov 9;64(1):35-41. doi: 10.1002/jmd2.12346. eCollection 2023 Jan. JIMD Rep. 2022. PMID: 36636599 Free PMC article.
-
A Rare Treatable Cause of Cardiomyopathy: Primary Carnitine Deficiency.Mol Syndromol. 2024 Mar;15(2):156-160. doi: 10.1159/000534932. Epub 2023 Nov 27. Mol Syndromol. 2024. PMID: 38585546 Free PMC article.
-
Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management.Orphanet J Rare Dis. 2012 Sep 18;7:68. doi: 10.1186/1750-1172-7-68. Orphanet J Rare Dis. 2012. PMID: 22989098 Free PMC article. Review.
-
Primary carnitine deficiency and cardiomyopathy.Korean Circ J. 2013 Dec;43(12):785-92. doi: 10.4070/kcj.2013.43.12.785. Korean Circ J. 2013. PMID: 24385988 Free PMC article. Review.
Cited by
-
A report of a pedigree with compound heterozygous mutations in the SLC22A5 gene.Front Pediatr. 2023 Jun 6;11:985720. doi: 10.3389/fped.2023.985720. eCollection 2023. Front Pediatr. 2023. PMID: 37351314 Free PMC article.
-
Screening primary carnitine deficiency in 10 million Chinese newborns: a systematic review and meta-analysis.Orphanet J Rare Dis. 2024 Jul 3;19(1):248. doi: 10.1186/s13023-024-03267-x. Orphanet J Rare Dis. 2024. PMID: 38961493 Free PMC article.
-
Clinical Efficacy and Safety of the Ketogenic Diet in Patients with Genetic Confirmation of Drug-Resistant Epilepsy.Nutrients. 2025 Mar 11;17(6):979. doi: 10.3390/nu17060979. Nutrients. 2025. PMID: 40290041 Free PMC article. Review.
-
The role of efflux transporters and metabolizing enzymes in brain and peripheral organs to explain drug-resistant epilepsy.Epilepsia Open. 2022 Aug;7 Suppl 1(Suppl 1):S47-S58. doi: 10.1002/epi4.12542. Epub 2021 Oct 1. Epilepsia Open. 2022. PMID: 34560816 Free PMC article. Review.
-
Clinical and Gene Analysis of Fatty Acid Oxidation Disorders Found in Neonatal Tandem Mass Spectrometry Screening.Pharmgenomics Pers Med. 2023 Jun 5;16:577-587. doi: 10.2147/PGPM.S402760. eCollection 2023. Pharmgenomics Pers Med. 2023. PMID: 37305019 Free PMC article.
References
-
- Greenberg DA, Aminoff MJ, Simon RP. Clinical Neurogy (5th edition). The United States: Mc Graw-Hill, New York, NY, pp157-158, 2002.
LinkOut - more resources
Full Text Sources