

Evolutionary trajectory of fish Piscine novirhabdovirus (=Viral Hemorrhagic Septicemia Virus) across its Laurentian Great Lakes history: Spatial and temporal diversification
- PMID: 33005343
- PMCID: PMC7520192
- DOI: 10.1002/ece3.6611
Evolutionary trajectory of fish Piscine novirhabdovirus (=Viral Hemorrhagic Septicemia Virus) across its Laurentian Great Lakes history: Spatial and temporal diversification
Abstract
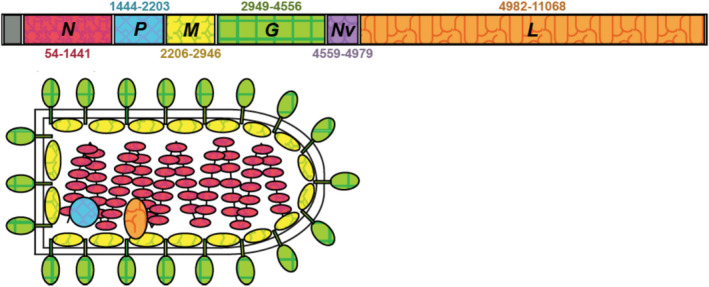
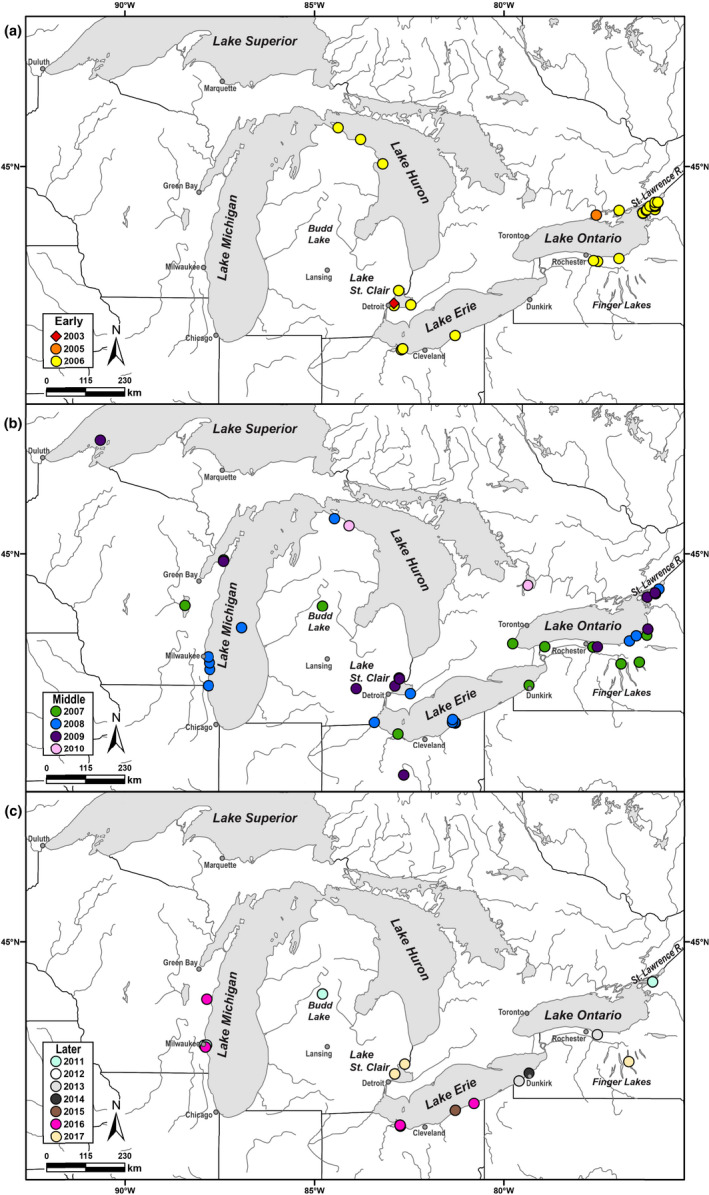
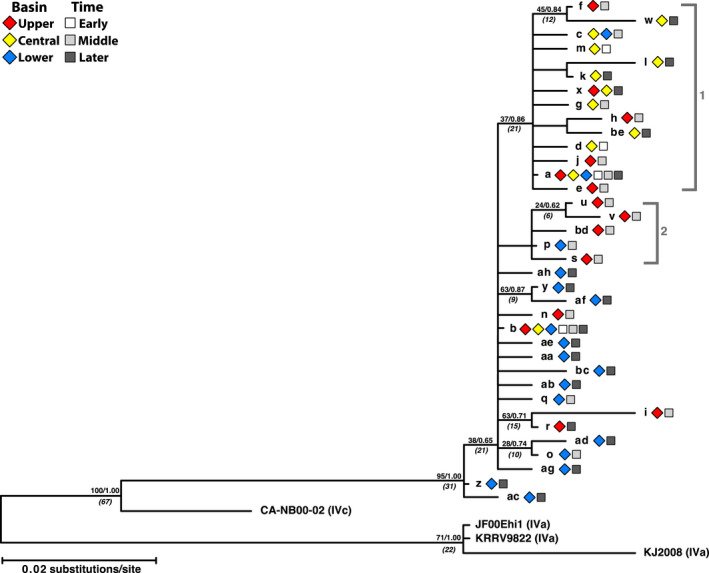
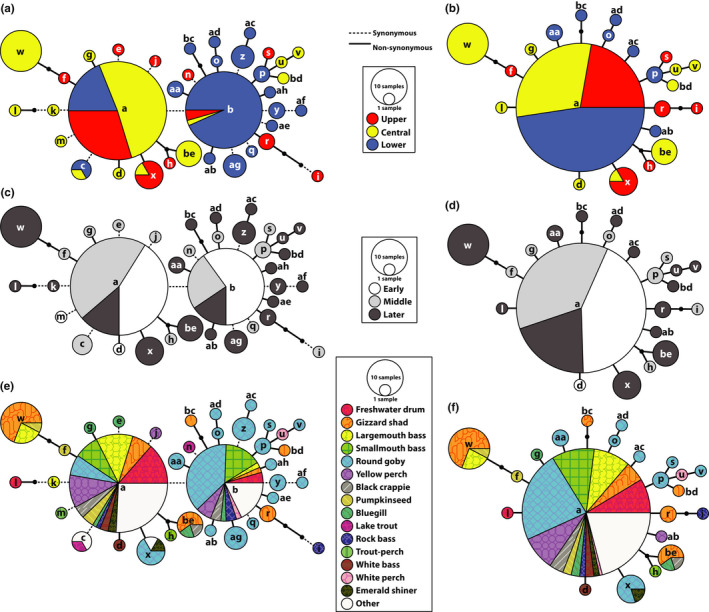
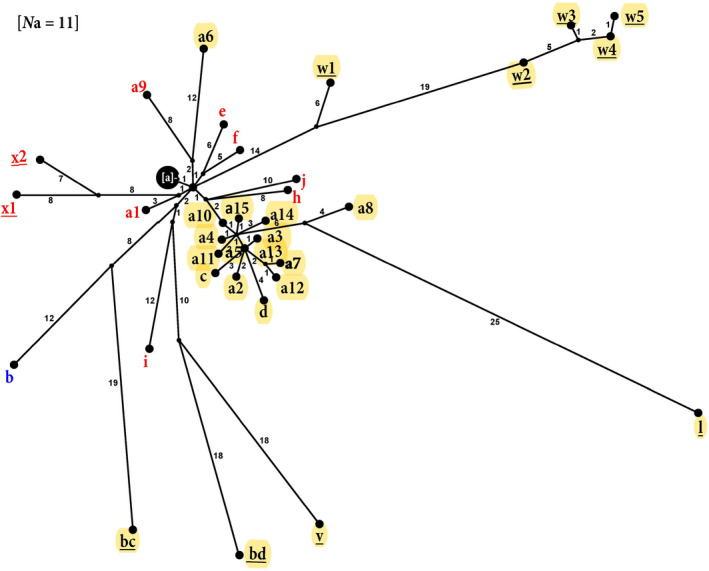
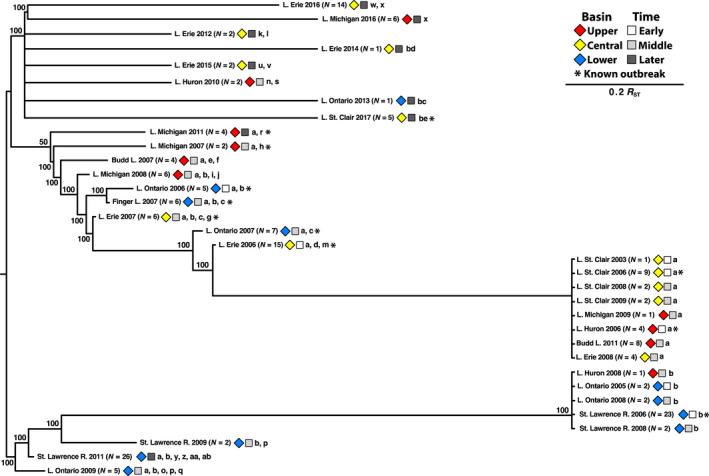
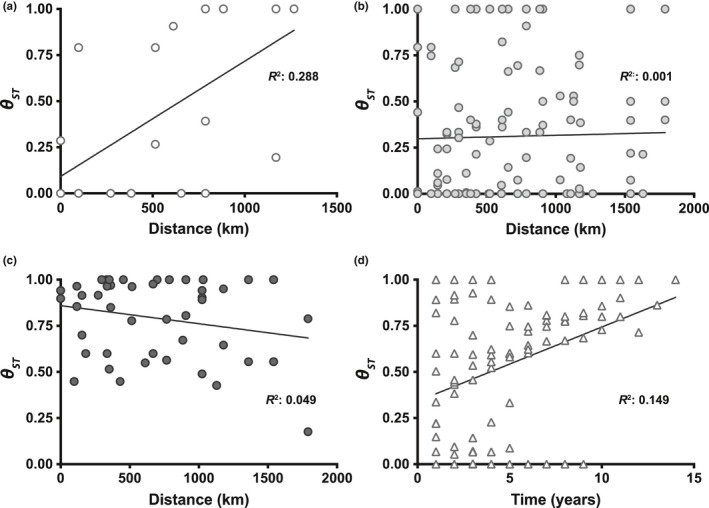
Piscine novirhabdovirus = Viral Hemorrhagic Septicemia Virus (VHSV) first appeared in the Laurentian Great Lakes with large outbreaks from 2005 to 2006, as a new and novel RNA rhabdovirus subgenogroup (IVb) that killed >30 fish species. Interlude periods punctuated smaller more localized outbreaks in 2007, 2010, and 2017, although some fishes tested positive in the intervals. There have not been reports of outbreaks or positives from 2018, 2019, or 2020. Here, we employ a combined population genetics and phylogenetic approach to evaluate spatial and temporal evolutionary trajectory on its G-gene sequence variation, in comparison with whole-genome sequences (11,083 bp) from a subset of 44 individual isolates (including 40 newly sequenced ones). Our results show that IVb (N = 184 individual fish isolates) diversified into 36 G-gene haplotypes from 2003 to 2017, stemming from two originals ("a" and "b"). G-gene haplotypes "a" and "b" differed by just one synonymous single-nucleotide polymorphism (SNP) substitution, remained the most abundant until 2011, then disappeared. Group "a" descendants (14 haplotypes) remained most prevalent in the Upper and Central Great Lakes, with eight (51%) having nonsynonymous substitutions. Group "b" descendants primarily have occurred in the Lower Great Lakes, including 22 haplotypes, of which 15 (68%) contained nonsynonymous changes. Evolutionary patterns of the whole-genome sequences (which had 34 haplotypes among 44 isolates) appear congruent with those from the G-gene. Virus populations significantly diverged among the Upper, Central, and Lower Great Lakes, diversifying over time. Spatial divergence was apparent in the overall patterns of nucleotide substitutions, while amino acid changes increased temporally. VHSV-IVb thus significantly differentiated across its less than two decades in the Great Lakes, accompanied by declining outbreaks and virulence. Continuing diversification likely allowed the virus to persist at low levels in resident fish populations, and may facilitate its potential for further and future spread to new habitats and nonacclimated hosts.
Keywords: RNA virus; Viral Hemorrhagic Septicemia Virus; evolutionary diversification; fish disease; novirhabdovirus; quasispecies; rhabdovirus.
© 2020 The Authors. Ecology and Evolution published by John Wiley & Sons Ltd.
Conflict of interest statement
None declared.
Figures
References
Associated data
LinkOut - more resources
Full Text Sources
