

Tissue Stem Cells: Architects of Their Niches
- PMID: 33007238
- PMCID: PMC7861346
- DOI: 10.1016/j.stem.2020.09.011
Tissue Stem Cells: Architects of Their Niches
Abstract
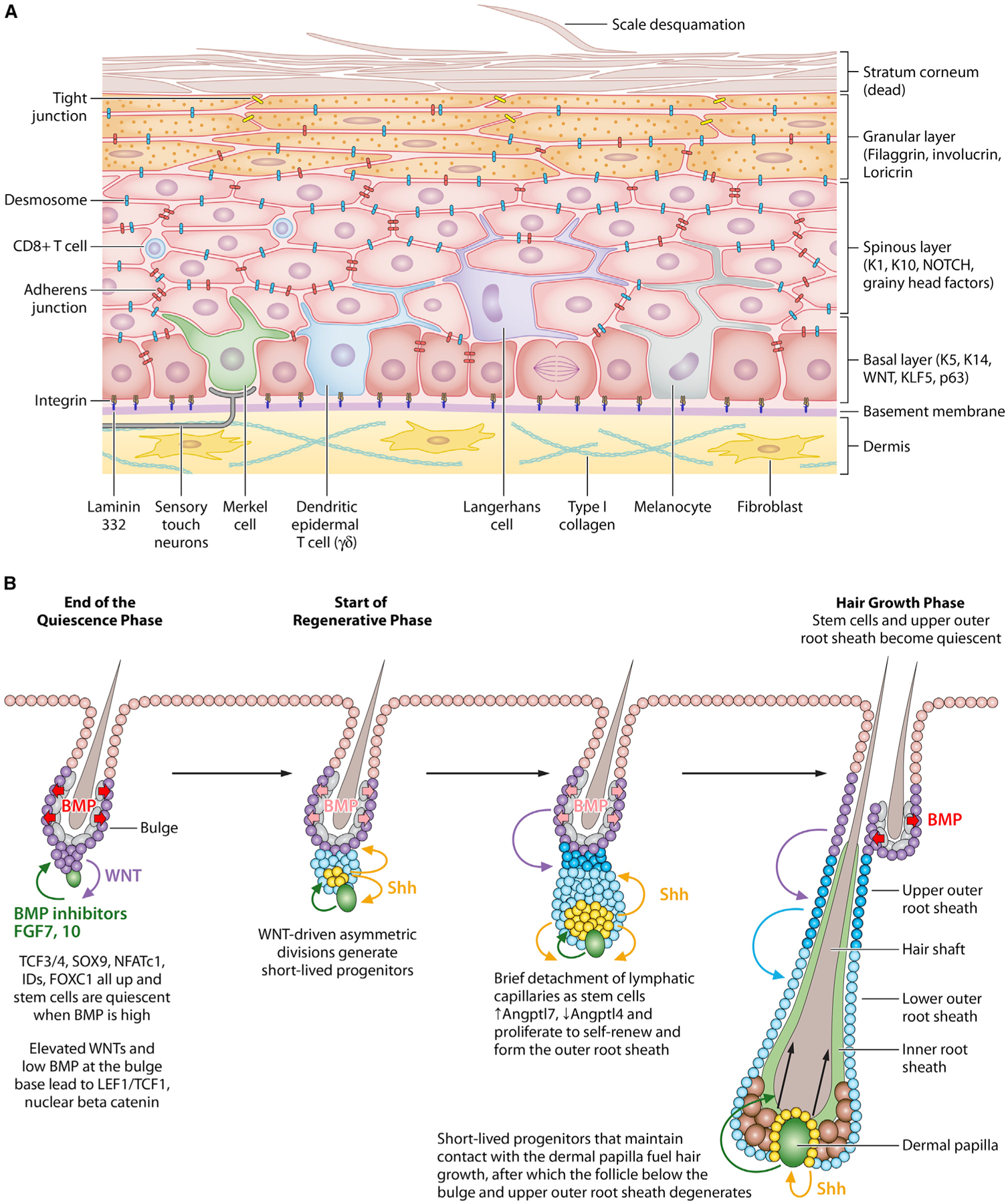
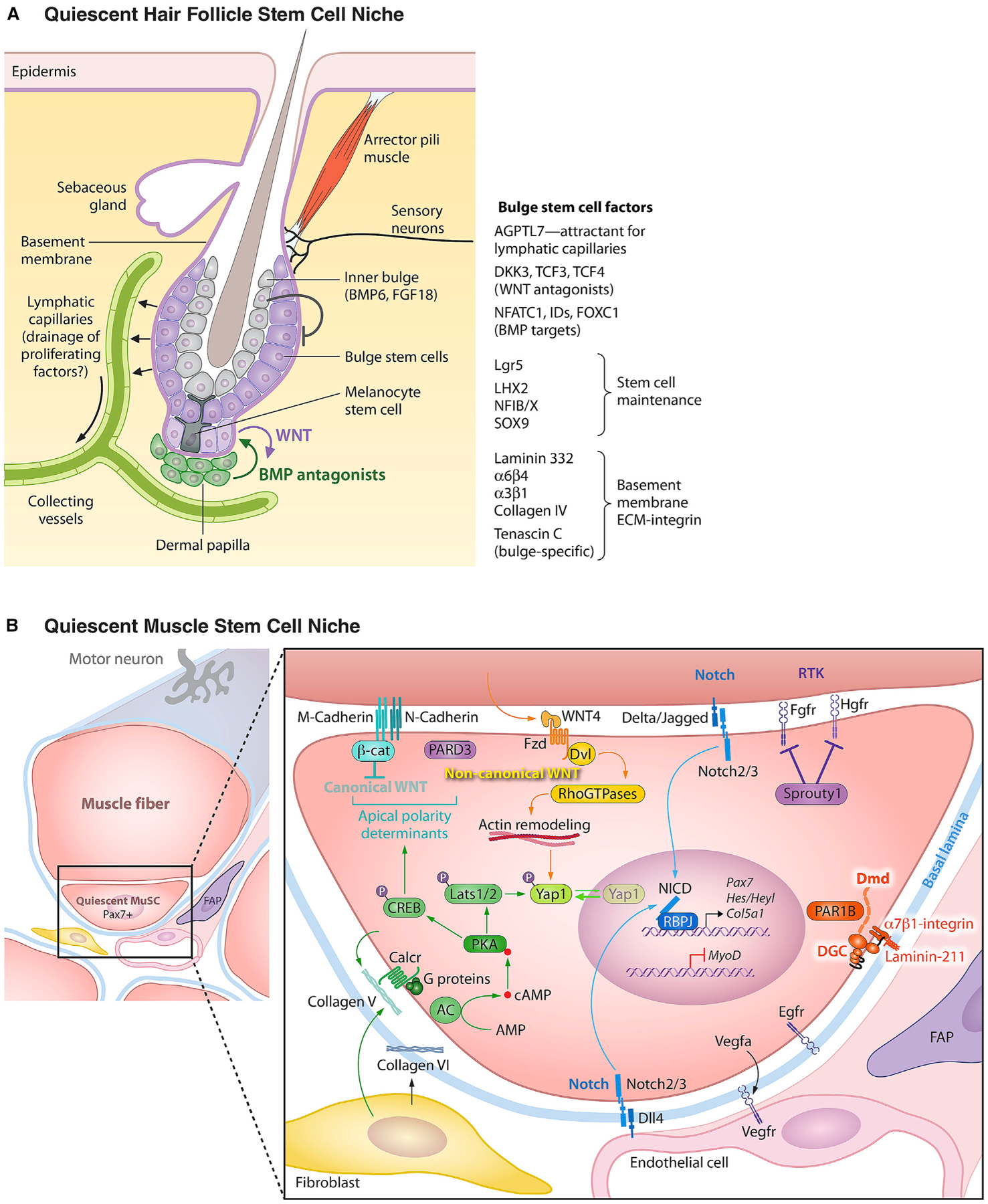
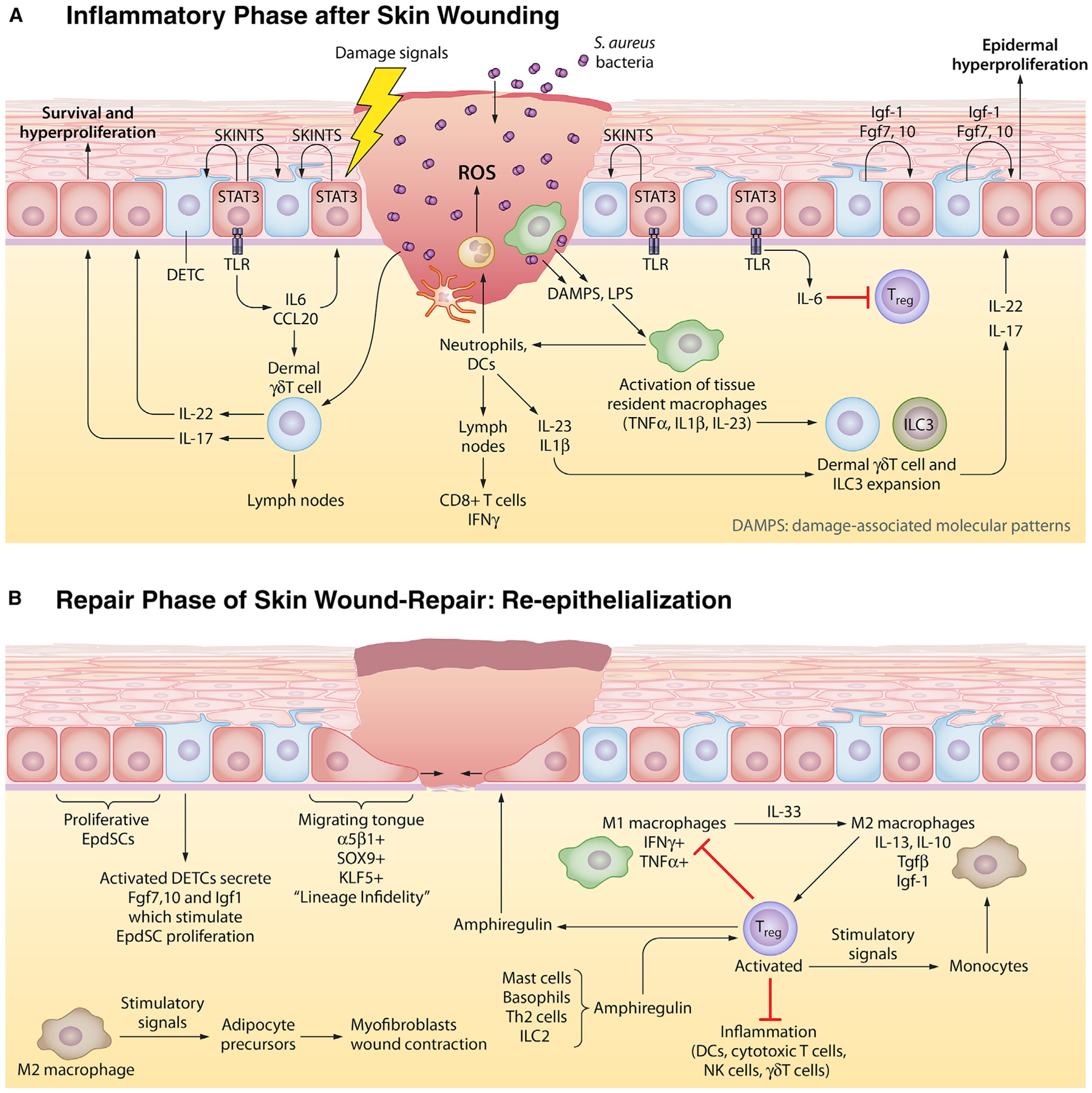
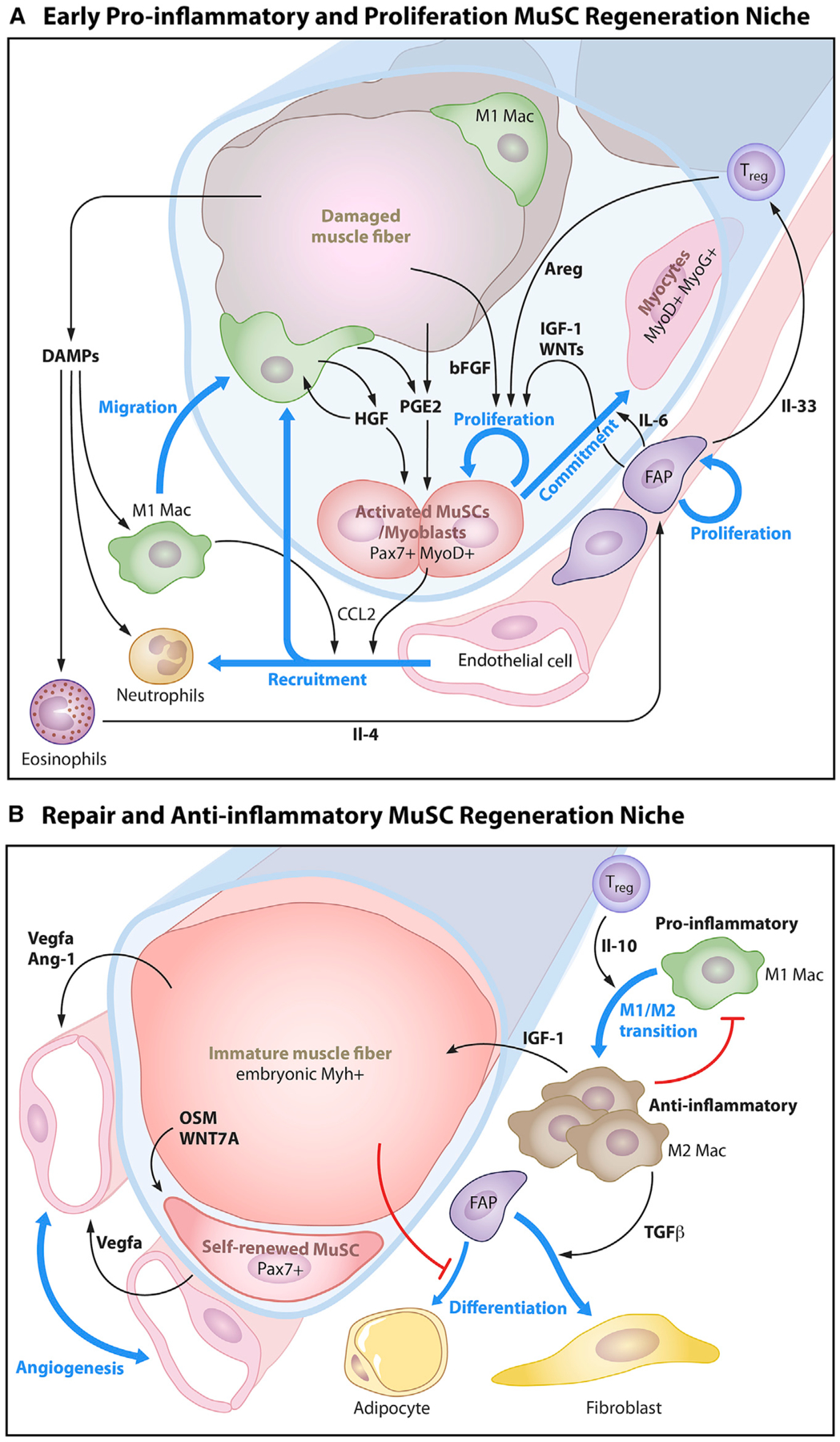
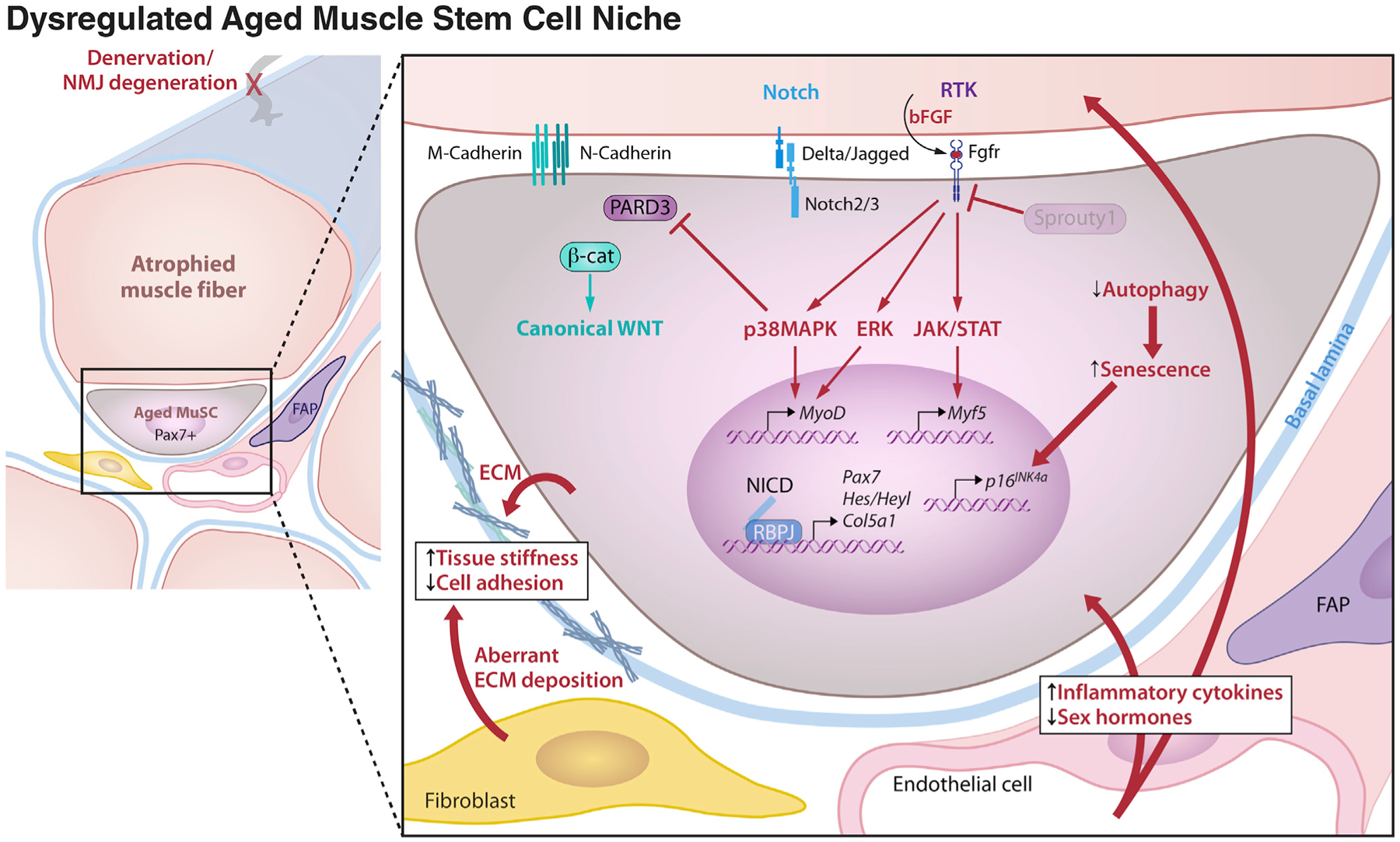
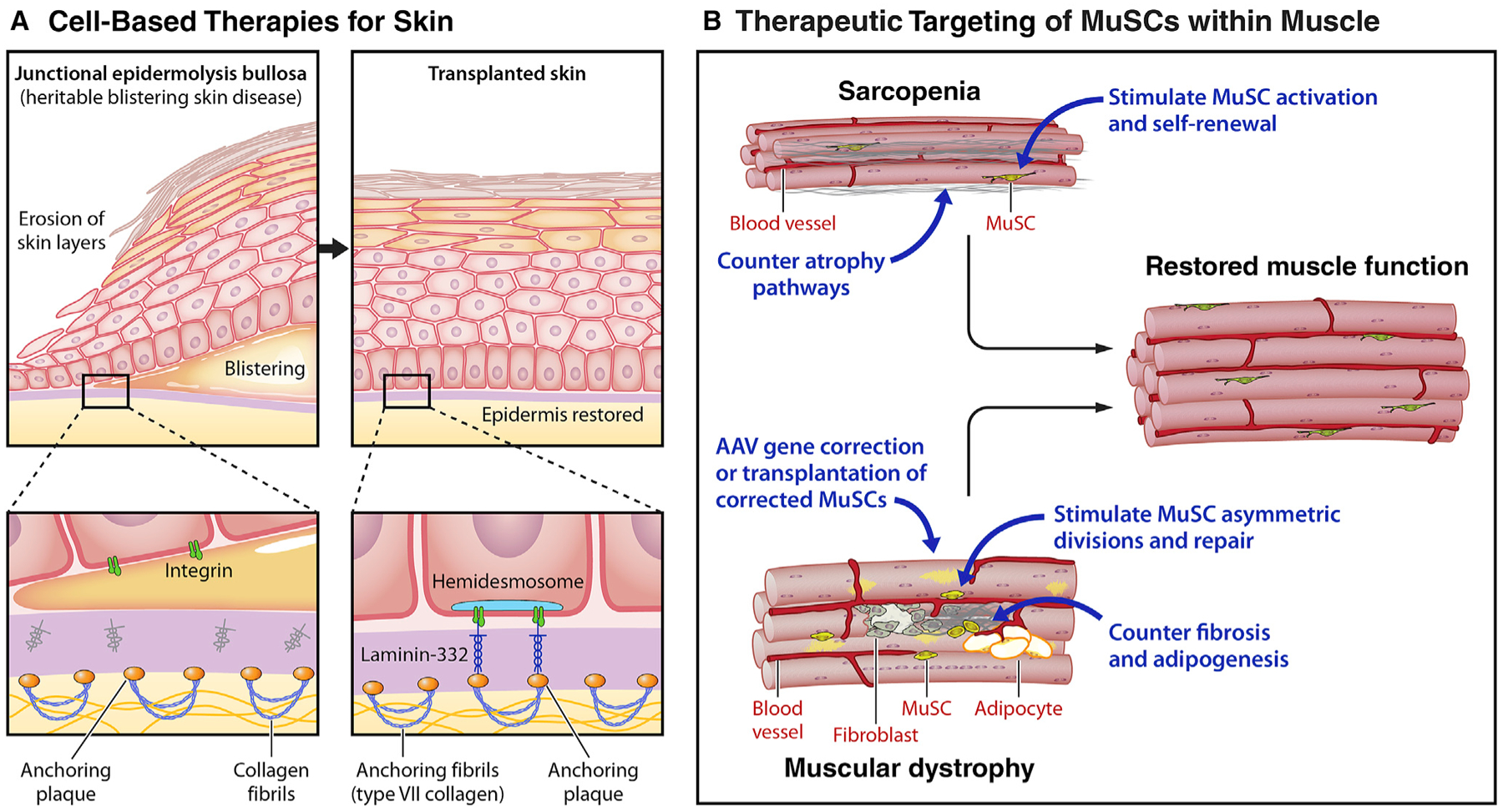
Stem cells (SCs) maintain tissue homeostasis and repair wounds. Despite marked variation in tissue architecture and regenerative demands, SCs often follow similar paradigms in communicating with their microenvironmental "niche" to transition between quiescent and regenerative states. Here we use skin epithelium and skeletal muscle-among the most highly-stressed tissues in our body-to highlight similarities and differences in niche constituents and how SCs mediate natural tissue rejuvenation and perform regenerative acts prompted by injuries. We discuss how these communication networks break down during aging and how understanding tissue SCs has led to major advances in regenerative medicine.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests E.F. is a member of the Scientific Advisory Boards of L’Oreal and Aresenal Biosciences. H.M.B. is a founder and consultant to Myoforte Therapeutics and a cofounder of Rejuvenation Technologies, and has two relevant issued patents. (1) Blau HM, Ho ATV, Palla A. Compositions and methods for muscle regeneration using prostaglandin E2. US9918994B1 3/20/18. (2) Ramunas, J., Yakubov, E, Blau, H.M., and Cooke, J. Compounds, compositions, methods, and kits relating to telomere extension US10525075B2 1/7/20.
Figures
References
-
- Anderson MS, and Kunkel LM (1992). The molecular and biochemical basis of Duchenne muscular dystrophy. Trends Biochem. Sci 17, 289–292. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
