

Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs
- PMID: 33007263
- PMCID: PMC7912537
- DOI: 10.1016/j.cell.2020.08.006
Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs
Abstract
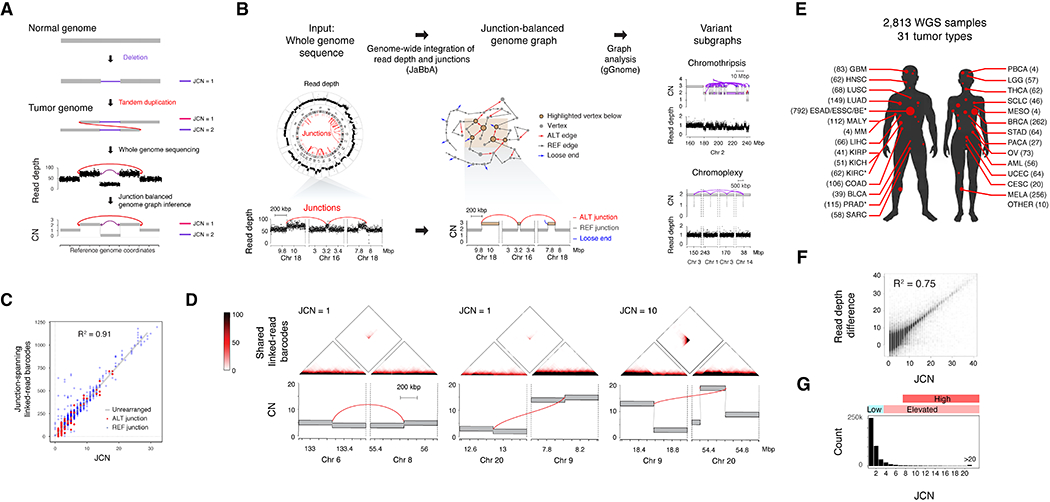
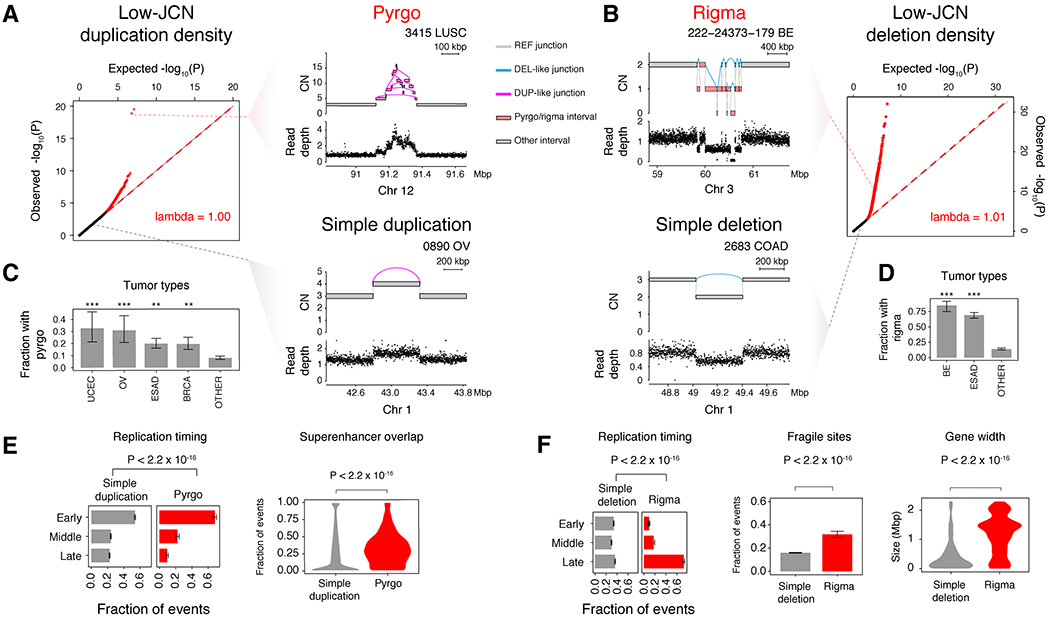
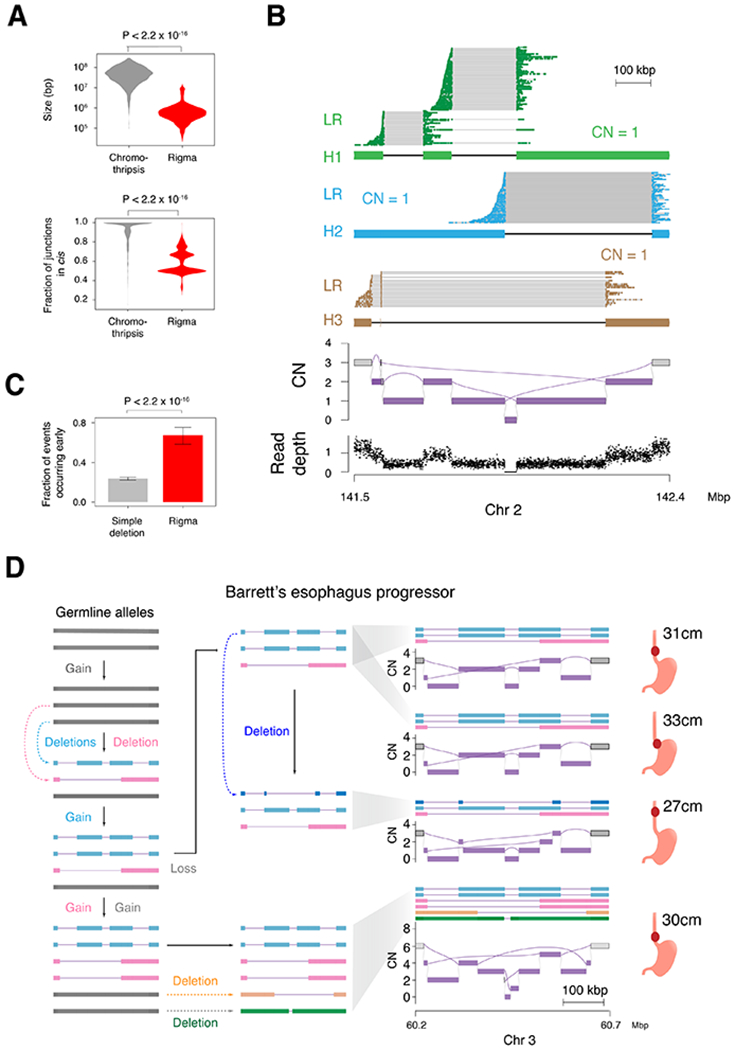
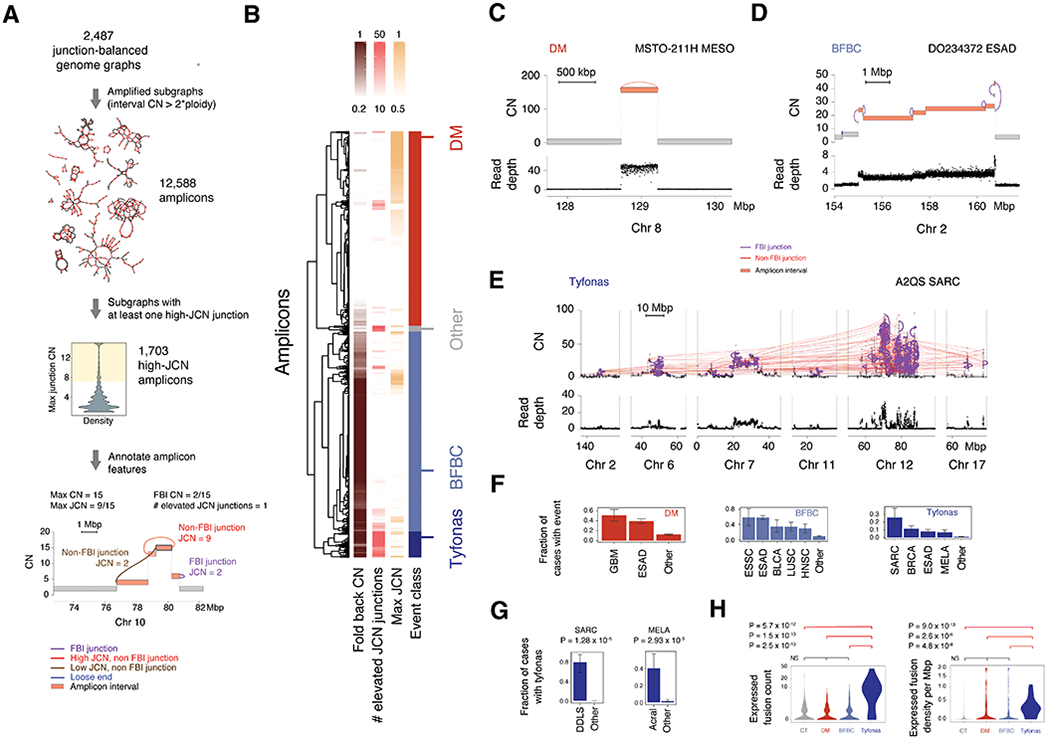
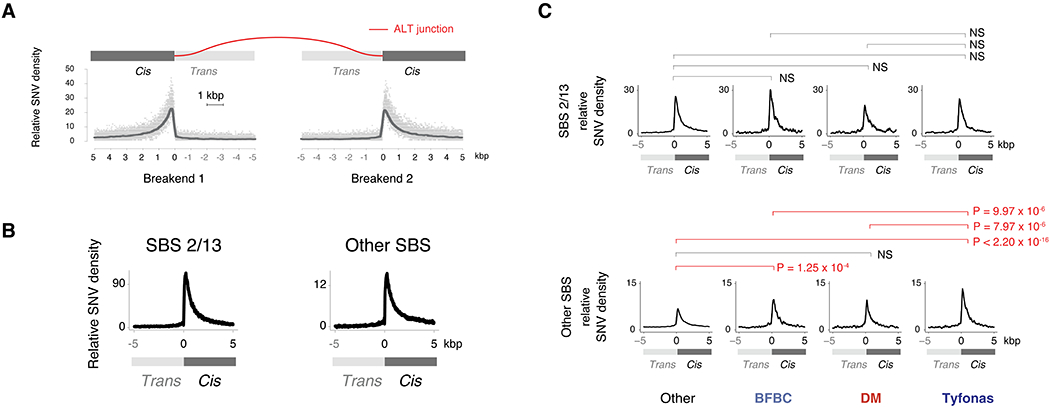
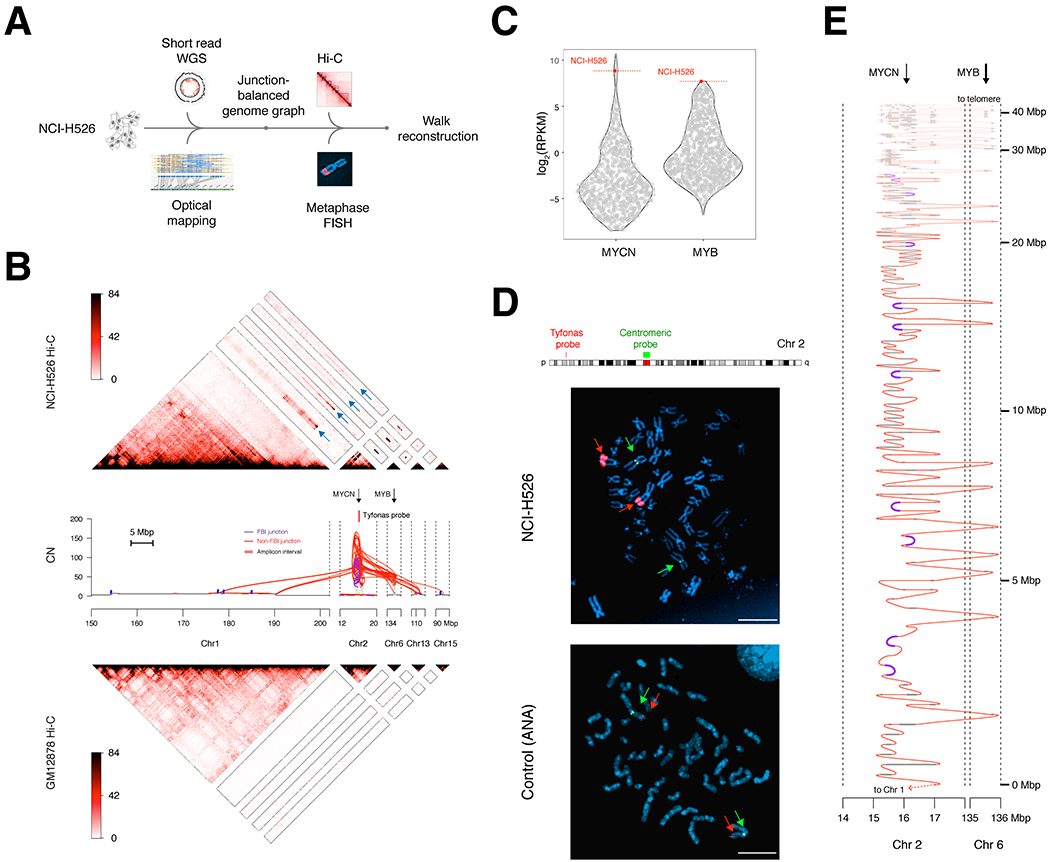
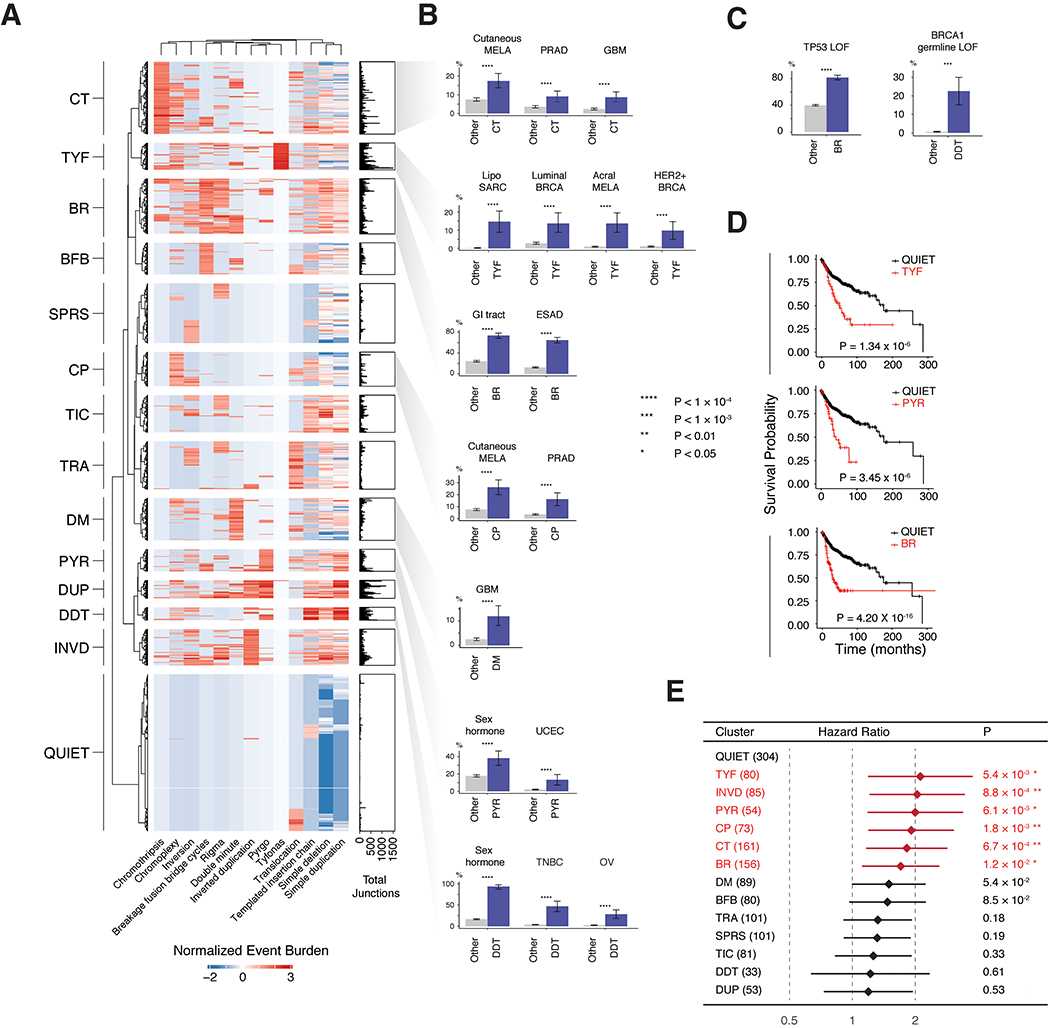
Cancer genomes often harbor hundreds of somatic DNA rearrangement junctions, many of which cannot be easily classified into simple (e.g., deletion) or complex (e.g., chromothripsis) structural variant classes. Applying a novel genome graph computational paradigm to analyze the topology of junction copy number (JCN) across 2,778 tumor whole-genome sequences, we uncovered three novel complex rearrangement phenomena: pyrgo, rigma, and tyfonas. Pyrgo are "towers" of low-JCN duplications associated with early-replicating regions, superenhancers, and breast or ovarian cancers. Rigma comprise "chasms" of low-JCN deletions enriched in late-replicating fragile sites and gastrointestinal carcinomas. Tyfonas are "typhoons" of high-JCN junctions and fold-back inversions associated with expressed protein-coding fusions, breakend hypermutation, and acral, but not cutaneous, melanomas. Clustering of tumors according to genome graph-derived features identified subgroups associated with DNA repair defects and poor prognosis.
Keywords: aneuploidy; cancer evolution; cancer genomics; chromothripsis; fragile sites; genome graphs; mutational processes; phasing; structural variation; superenhancers.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests J.S.R.-F. reports receiving personal/consultancy fees from VolitionRx, Paige.AI, Goldman Sachs, REPARE Therapeutics, GRAIL, Ventana Medical Systems, Roche, Genentech, and InviCRO outside of the scope of the submitted work.
Figures
References
-
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, PCAWG Mutational Signatures Working Group, Getz G, Rozen SG, Stratton MR. and PCAWG Consortium (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. - PMC - PubMed
-
- Anantharaman T and Mishra B (2001). False positives in genomic map assembly and sequence validation. In International Workshop on Algorithms in Bioinformatics 27–40, Springer.
-
- Baca S, Prandi D, Lawrence M, Mosquera J, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald T, Ghandi M, Van Allen E, Kryukov G, Sboner A, Theurillat J-P, Soong T, Nickerson E, Auclair D, Tewari A, Beltran H, Onofrio R, Boysen G, Guiducci C, Barbieri C, Cibulskis K, Sivachenko A, Carter S, Saksena G, Voet D, Ramos A, Winckler W, Cipicchio M, Ardlie K, Kantoff P, Berger M, Gabriel S, Golub T, Meyerson M, Lander E, Elemento O , Getz G, Demichelis F, Rubin M and Garraway L (2013). Punctuated Evolution of Prostate Cancer Genomes. Cell. 153, 666–677. - PMC - PubMed
-
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr , de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R and Garraway LA. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. - PMC - PubMed
-
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Henry KTM, Pinchback RM, Ligon AH, Cho Y-J, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q , Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao M-S, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR and Meyerson M (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- UL1 TR000043/TR/NCATS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- P01 CA091955/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R35 NS097404/NS/NINDS NIH HHS/United States
- P50 CA247749/CA/NCI NIH HHS/United States
- U24 CA210989/CA/NCI NIH HHS/United States
- P30 CA015704/CA/NCI NIH HHS/United States
- U54 CA193313/CA/NCI NIH HHS/United States
- R01 CA194547/CA/NCI NIH HHS/United States
- F31 CA232465/CA/NCI NIH HHS/United States
- T32 GM132083/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
