

BacWGSTdb 2.0: a one-stop repository for bacterial whole-genome sequence typing and source tracking
- PMID: 33010178
- PMCID: PMC7778894
- DOI: 10.1093/nar/gkaa821
BacWGSTdb 2.0: a one-stop repository for bacterial whole-genome sequence typing and source tracking
Abstract
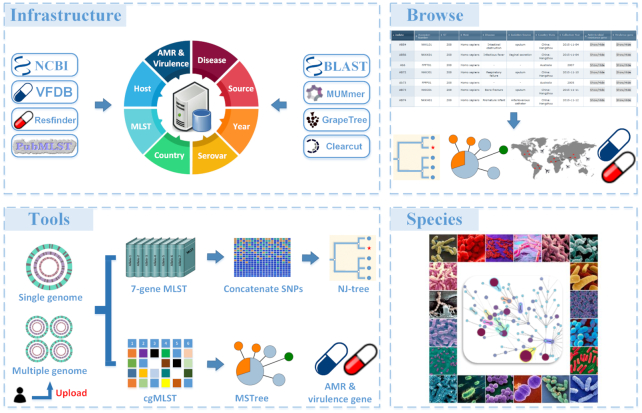
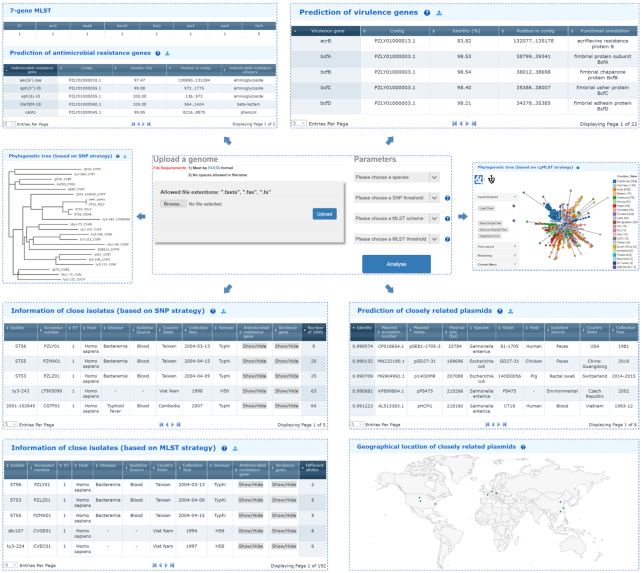
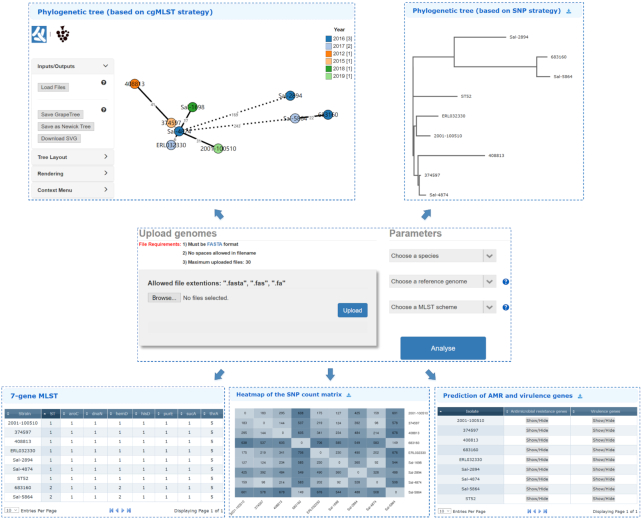
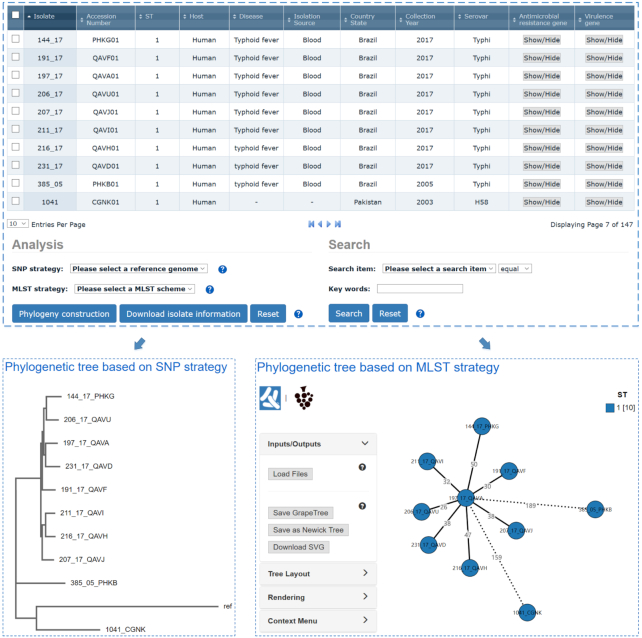
An increasing prevalence of hospital acquired infections and foodborne illnesses caused by pathogenic and multidrug-resistant bacteria has stimulated a pressing need for benchtop computational techniques to rapidly and accurately classify bacteria from genomic sequence data, and based on that, to trace the source of infection. BacWGSTdb (http://bacdb.org/BacWGSTdb) is a free publicly accessible database we have developed for bacterial whole-genome sequence typing and source tracking. This database incorporates extensive resources for bacterial genome sequencing data and the corresponding metadata, combined with specialized bioinformatics tools that enable the systematic characterization of the bacterial isolates recovered from infections. Here, we present BacWGSTdb 2.0, which encompasses several major updates, including (i) the integration of the core genome multi-locus sequence typing (cgMLST) approach, which is highly scalable and appropriate for typing isolates belonging to different lineages; (ii) the addition of a multiple genome analysis module that can process dozens of user uploaded sequences in a batch mode; (iii) a new source tracking module for comparing user uploaded plasmid sequences to those deposited in the public databases; (iv) the number of species encompassed in BacWGSTdb 2.0 has increased from 9 to 20, which represents bacterial pathogens of medical importance; (v) a newly designed, user-friendly interface and a set of visualization tools for providing a convenient platform for users are also included. Overall, the updated BacWGSTdb 2.0 bears great utility in continuing to provide users, including epidemiologists, clinicians and bench scientists, with a one-stop solution to bacterial genome sequence analysis.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Brachman P.S. Infectious diseases–past, present, and future. Int. J. Epidemiol. 2003; 32:684–686. - PubMed
-
- Hernando-Amado S., Coque T.M., Baquero F., Martinez J.L.. Defining and combating antibiotic resistance from One Health and Global Health perspectives. Nat. Microbiol. 2019; 4:1432–1442. - PubMed
-
- Scott P., Deye G., Srinivasan A., Murray C., Moran K., Hulten E., Fishbain J., Craft D., Riddell S., Lindler L. et al.. An outbreak of multidrug-resistant Acinetobacter baumannii-calcoaceticus complex infection in the US military health care system associated with military operations in Iraq. Clin. Infect. Dis. 2007; 44:1577–1584. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
