

Necrosis-Released HMGB1 (High Mobility Group Box 1) in the Progressive Pulmonary Arterial Hypertension Associated With Male Sex
- PMID: 33012199
- PMCID: PMC7666015
- DOI: 10.1161/HYPERTENSIONAHA.120.16118
Necrosis-Released HMGB1 (High Mobility Group Box 1) in the Progressive Pulmonary Arterial Hypertension Associated With Male Sex
Abstract
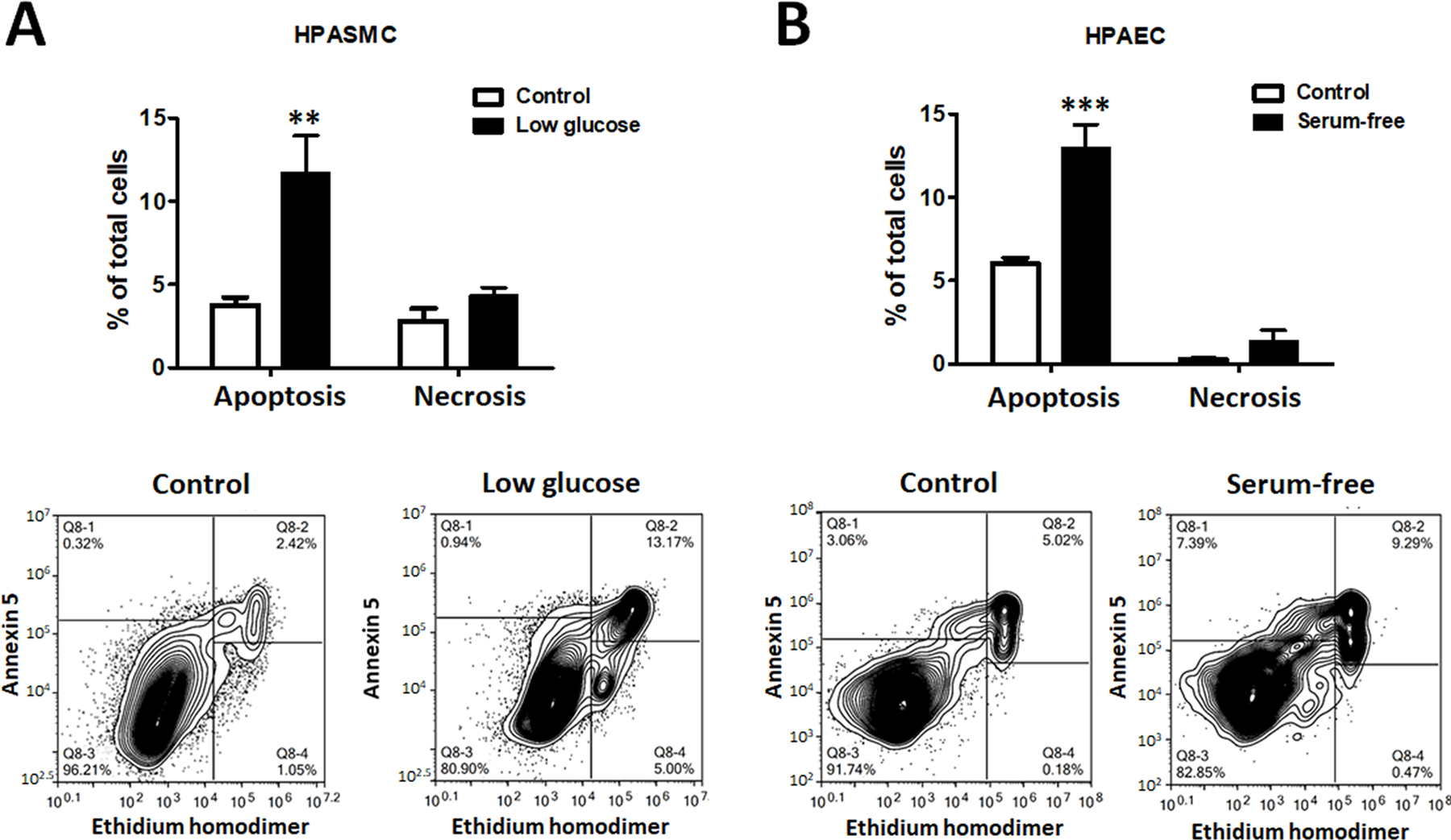
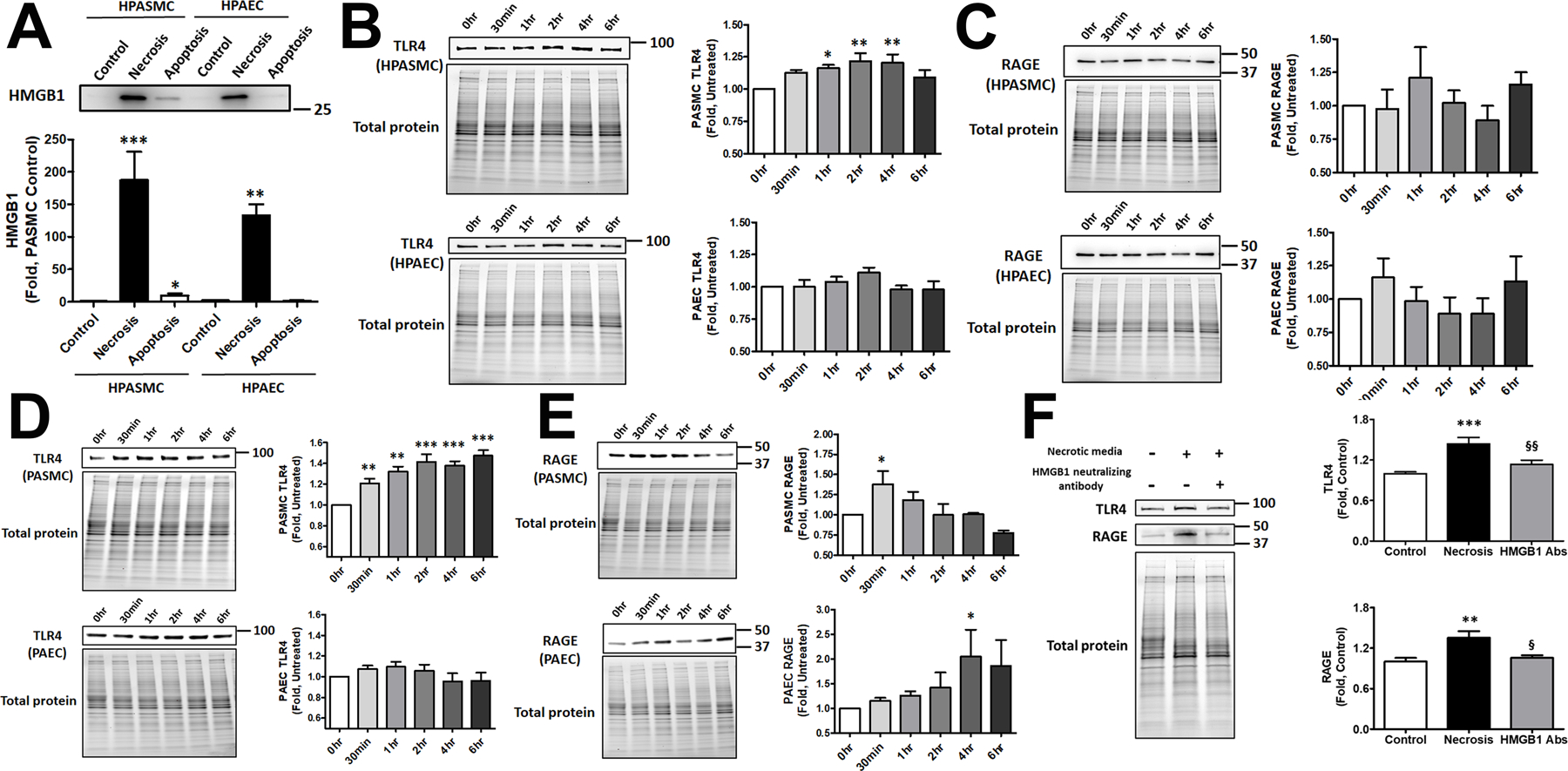
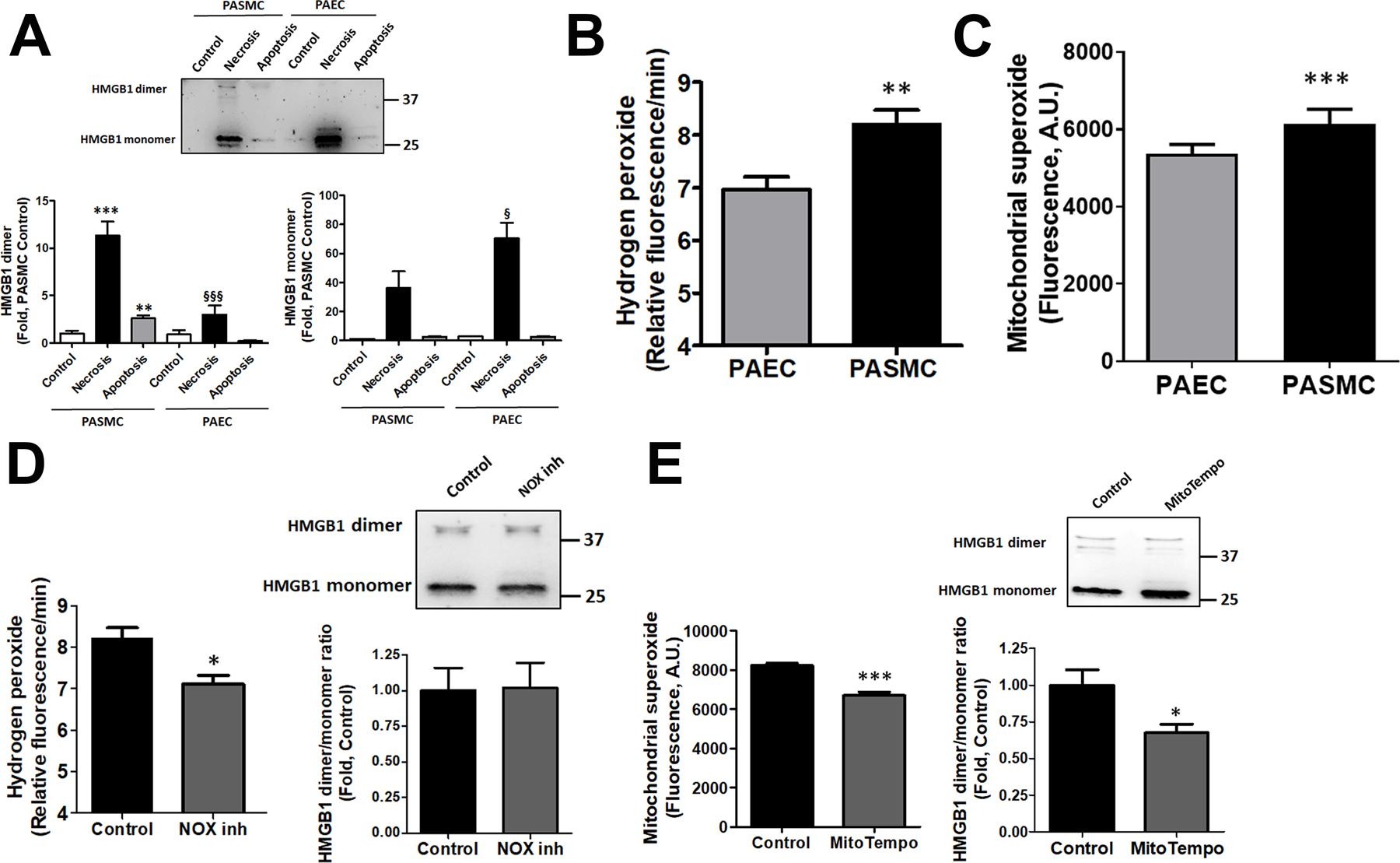
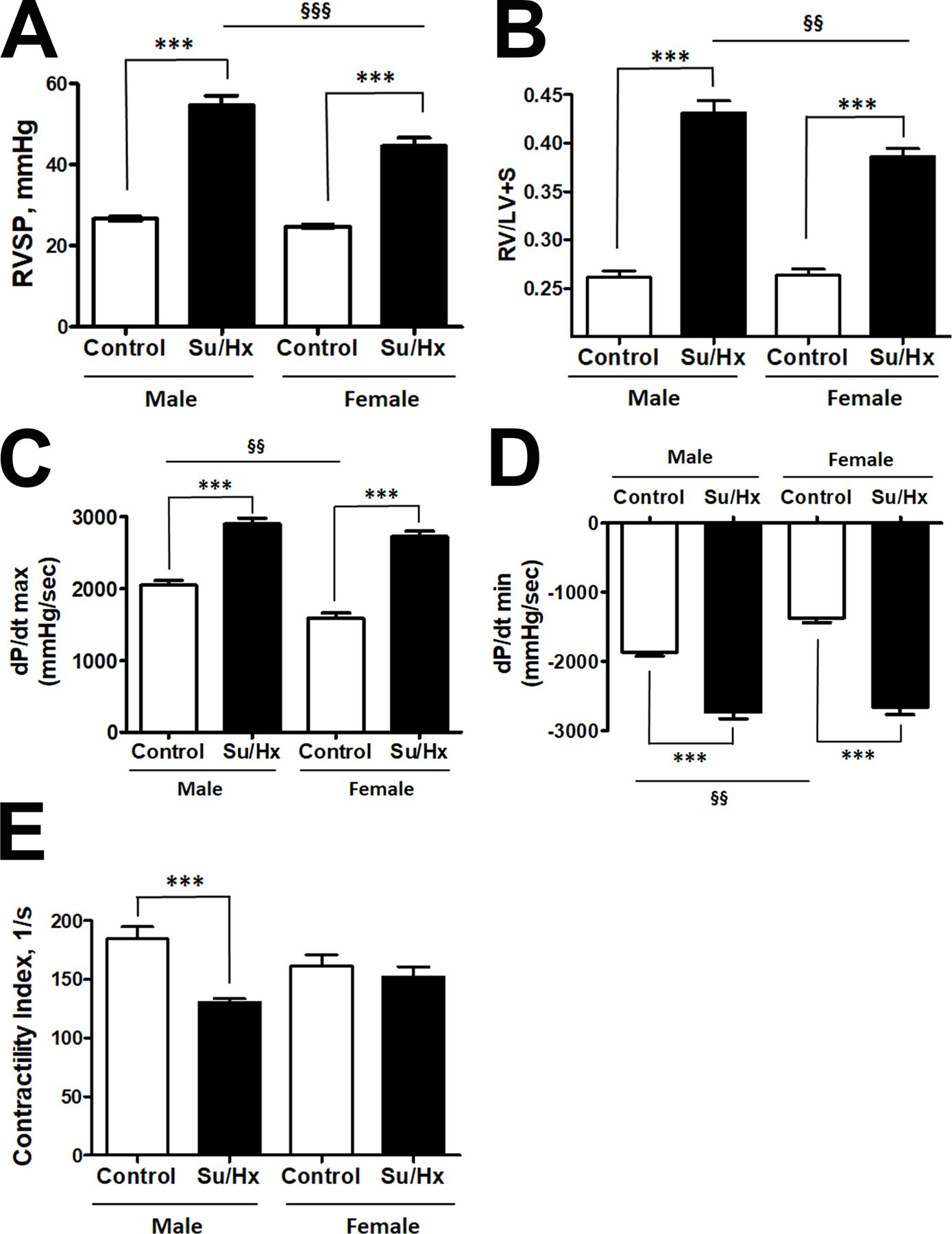
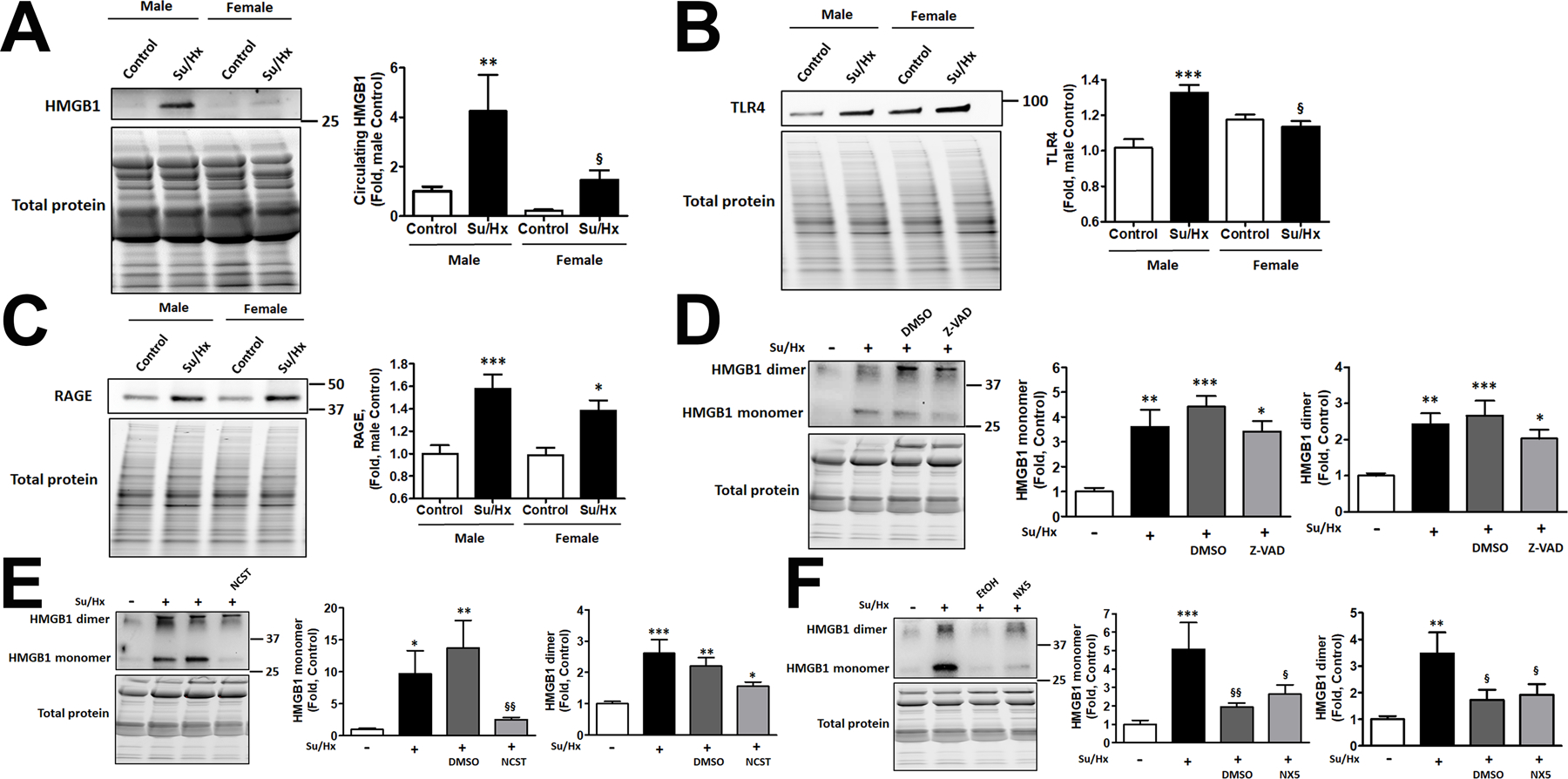
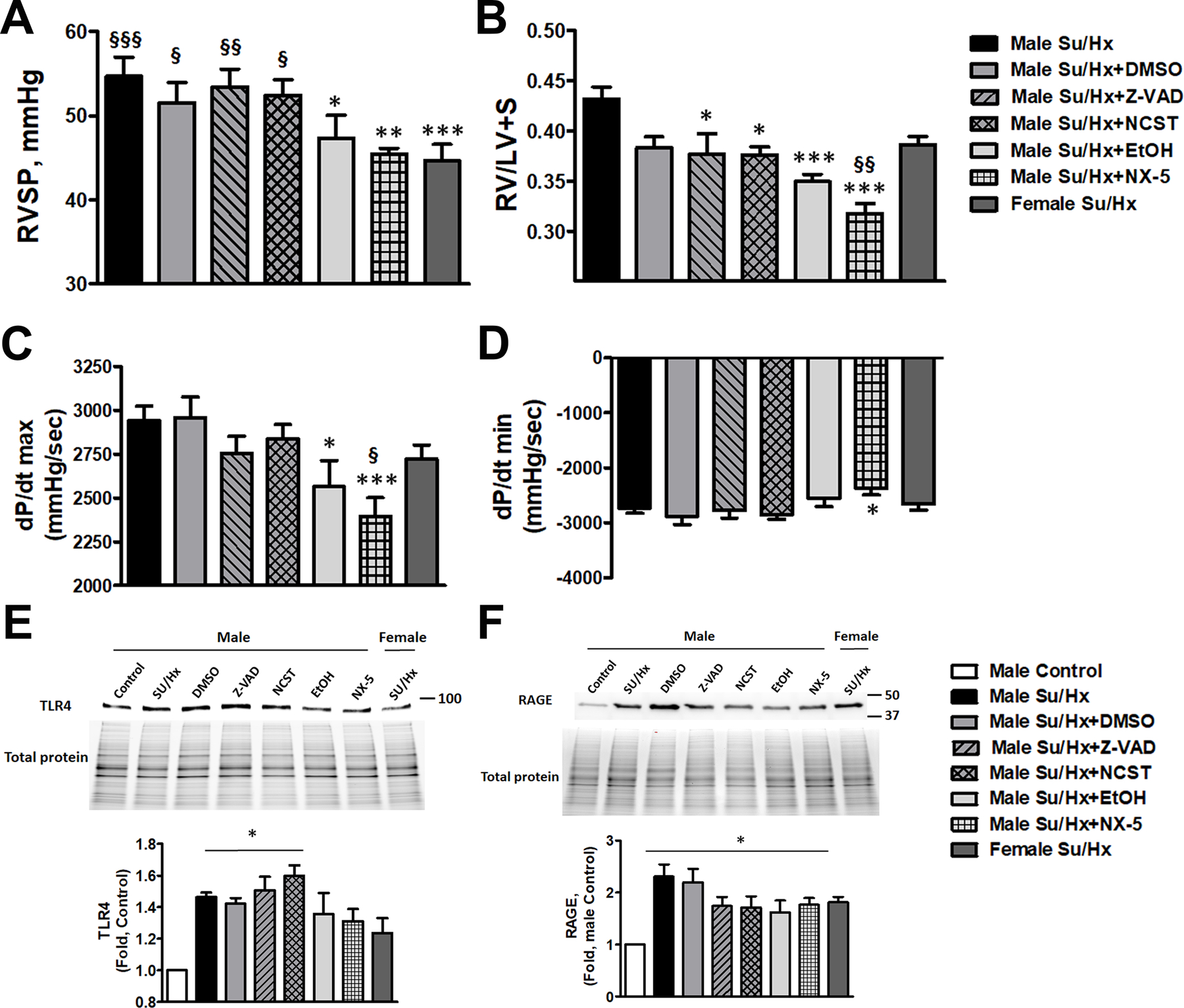
Damage-associated molecular patterns, such as HMGB1 (high mobility group box 1), play a well-recognized role in the development of pulmonary arterial hypertension (PAH), a progressive fatal disease of the pulmonary vasculature. However, the contribution of the particular type of vascular cells, type of cell death, or the form of released HMGB1 in PAH remains unclear. Moreover, although male patients with PAH show a higher level of circulating HMGB1, its involvement in the severe PAH phenotype reported in males is unknown. In this study, we aimed to investigate the sources and active forms of HMGB1 released from damaged vascular cells and their contribution to the progressive type of PAH in males. Our results showed that HMGB1 is released by either pulmonary artery human endothelial cells or human pulmonary artery smooth muscle cells that underwent necrotic cell death, although only human pulmonary artery smooth muscle cells produce HMGB1 during apoptosis. Moreover, only human pulmonary artery smooth muscle cell death induced a release of dimeric HMGB1, found to be mitochondrial reactive oxygen species dependent, and TLR4 (toll-like receptor 4) activation. The modified Sugen/Hypoxia rat model replicates the human sexual dimorphism in PAH severity (right ventricle systolic pressure in males versus females 54.7±2.3 versus 44.6±2 mm Hg). By using this model, we confirmed that necroptosis and necrosis are the primary sources of circulating HMGB1 in the male rats, although only necrosis increased circulation of HMGB1 dimers. Attenuation of necrosis but not apoptosis or necroptosis prevented TLR4 activation in males and blunted the sex differences in PAH severity. We conclude that necrosis, through the release of HMGB1 dimers, predisposes males to a progressive form of PAH.
Keywords: apoptosis; hypertension, pulmonary; hypoxia; necrosis; phenotype.
Figures
Similar articles
-
Gender Difference in Damage-Mediated Signaling Contributes to Pulmonary Arterial Hypertension.Antioxid Redox Signal. 2019 Nov 1;31(13):917-932. doi: 10.1089/ars.2018.7664. Epub 2019 Mar 20. Antioxid Redox Signal. 2019. PMID: 30652485 Free PMC article.
-
HMGB1/TLR4 promotes hypoxic pulmonary hypertension via suppressing BMPR2 signaling.Vascul Pharmacol. 2019 Jun;117:35-44. doi: 10.1016/j.vph.2018.12.006. Epub 2019 Jan 3. Vascul Pharmacol. 2019. PMID: 30610955
-
The Critical Role of HMGB1Cys106 in Regulating Sex-Specific p53 Signaling in Pulmonary Arterial Hypertension.Am J Respir Cell Mol Biol. 2025 Sep;73(3):396-414. doi: 10.1165/rcmb.2024-0296OC. Am J Respir Cell Mol Biol. 2025. PMID: 40085493
-
Phenotypic Diversity of Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension: Implications for Therapy.Chest. 2022 Jan;161(1):219-231. doi: 10.1016/j.chest.2021.08.040. Epub 2021 Aug 12. Chest. 2022. PMID: 34391758 Review.
-
Panorama of artery endothelial cell dysfunction in pulmonary arterial hypertension.J Mol Cell Cardiol. 2024 Dec;197:61-77. doi: 10.1016/j.yjmcc.2024.10.004. Epub 2024 Oct 20. J Mol Cell Cardiol. 2024. PMID: 39437884 Review.
Cited by
-
Toll-Like Receptors Represent an Important Link for Sex Differences in Cardiovascular Aging and Diseases.Front Aging. 2021 Jun 24;2:709914. doi: 10.3389/fragi.2021.709914. eCollection 2021. Front Aging. 2021. PMID: 35822020 Free PMC article. Review.
-
Exploring the diagnostic and immune infiltration roles of disulfidptosis related genes in pulmonary hypertension.Respir Res. 2024 Oct 9;25(1):365. doi: 10.1186/s12931-024-02978-w. Respir Res. 2024. PMID: 39385167 Free PMC article.
-
The mechanism of programmed death and endoplasmic reticulum stress in pulmonary hypertension.Cell Death Discov. 2023 Feb 25;9(1):78. doi: 10.1038/s41420-023-01373-6. Cell Death Discov. 2023. PMID: 36841823 Free PMC article. Review.
-
Necroptosis in Pulmonary Diseases: A New Therapeutic Target.Front Pharmacol. 2021 Sep 14;12:737129. doi: 10.3389/fphar.2021.737129. eCollection 2021. Front Pharmacol. 2021. PMID: 34594225 Free PMC article. Review.
-
New insights into pulmonary arterial hypertension: interaction between PANoptosis and perivascular inflammatory responses.Apoptosis. 2025 Jun;30(5-6):1097-1116. doi: 10.1007/s10495-025-02086-0. Epub 2025 Feb 20. Apoptosis. 2025. PMID: 39979525 Free PMC article. Review.
References
-
- Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF and Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:427–38. - PubMed
-
- Jurasz P, Courtman D, Babaie S and Stewart DJ. Role of apoptosis in pulmonary hypertension: from experimental models to clinical trials. Pharmacology & therapeutics. 2010;126:1–8. - PubMed
-
- Levy M, Maurey C, Celermajer DS, Vouhe PR, Danel C, Bonnet D and Israel-Biet D. Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. Journal of the American College of Cardiology. 2007;49:803–10. - PubMed
-
- Heath D and Whitaker W. Hypertensive pulmonary vascular disease. Circulation. 1956;14:323–43. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
