

Superoxide and Non-ionotropic Signaling in Neuronal Excitotoxicity
- PMID: 33013314
- PMCID: PMC7497801
- DOI: 10.3389/fnins.2020.00861
Superoxide and Non-ionotropic Signaling in Neuronal Excitotoxicity
Abstract
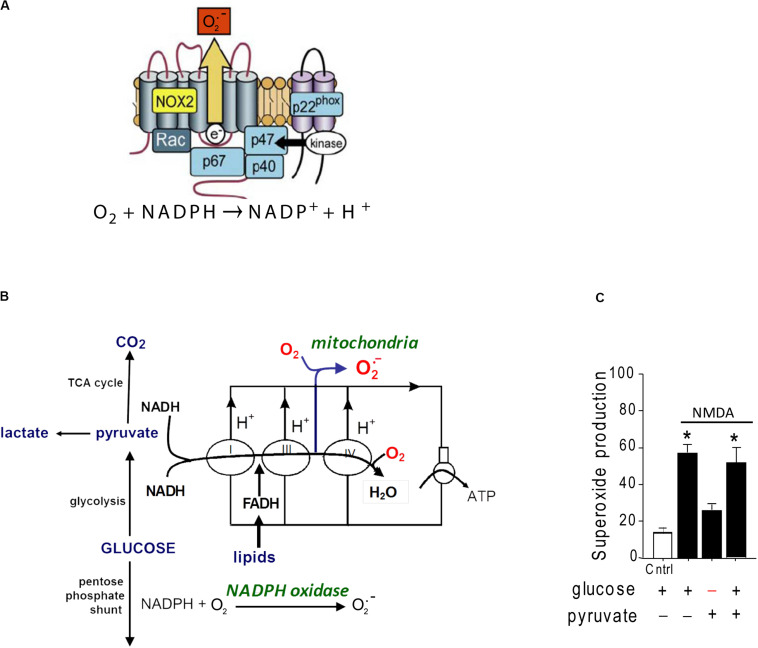
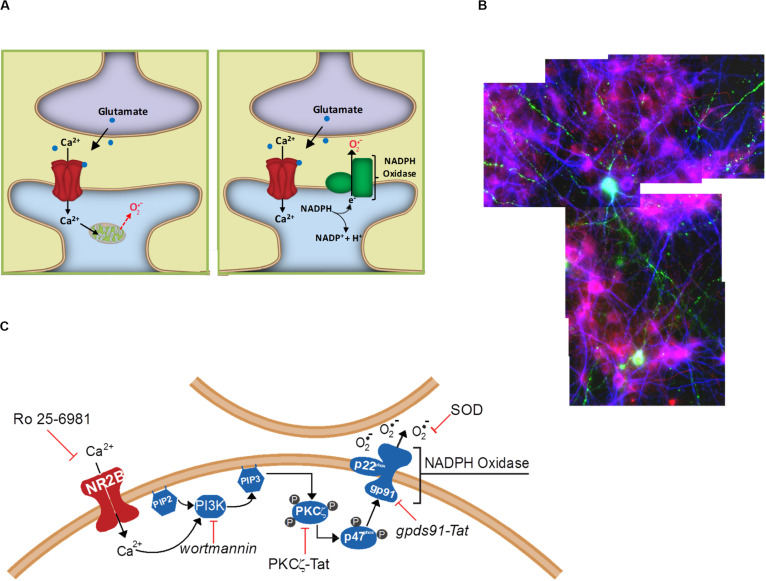
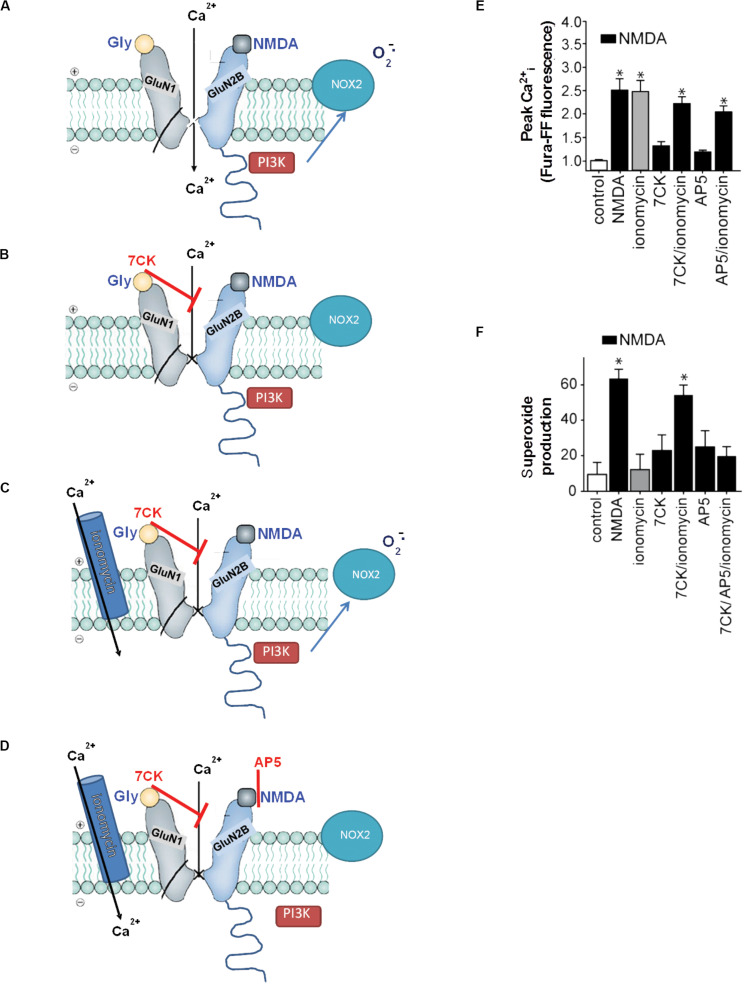
Excitotoxicity is classically attributed to Ca2+ influx through NMDA receptors (NMDAr), leading to production of nitric oxide by neuronal nitric oxide synthase and superoxide by mitochondria, which react to form highly cytotoxic peroxynitrite. More recent observations warrant revision of the classic view and help to explain some otherwise puzzling aspects of excitotoxic cell injury. Studies using pharmacological and genetic approaches show that superoxide produced by NMDAr activation originates primarily from NADPH oxidase rather than from mitochondria. As NADPH oxidase is localized to the plasma membrane, this also provides an explanation for the extracellular release of superoxide and cell-to-cell "spread" of excitotoxic injury observed in vitro and in vivo. The signaling pathway linking NMDAr to NADPH oxidase involves Ca2+ influx, phosphoinositol-3-kinase, and protein kinase Cζ, and interventions at any of these steps can prevent superoxide production and excitotoxic injury. Ca2+ influx specifically through NMDAr is normally required to induce excitotoxicity, through a mechanism presumed to involve privileged Ca2+ access to local signaling domains. However, experiments using selective blockade of the NMDAr ion channel and artificial reconstitution of Ca2+ by other routes indicate that the special effects of NMDAr activation are attributable instead to concurrent non-ionotropic NMDAr signaling by agonist binding to NMDAr. The non-ionotropic signaling driving NADPH oxidase activation is mediated in part by phosphoinositol-3-kinase binding to the C-terminal domain of GluN2B receptor subunits. These more recently identified aspects of excitotoxicity expand our appreciation of the complexity of excitotoxic processes and suggest novel approaches for limiting neuronal injury.
Keywords: Glun2B; NADPH oxidase; calcium; glucose; glutamate; metabotropic; peroxynitrite; phosphoinositol-3-kinase.
Copyright © 2020 Wang and Swanson.
Figures
References
-
- Andreyev A. Y., Kushnareva Y. E., Starkov A. A. (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70 200–214. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous