

Computational Methods for Strain-Level Microbial Detection in Colony and Metagenome Sequencing Data
- PMID: 33013732
- PMCID: PMC7507117
- DOI: 10.3389/fmicb.2020.01925
Computational Methods for Strain-Level Microbial Detection in Colony and Metagenome Sequencing Data
Abstract
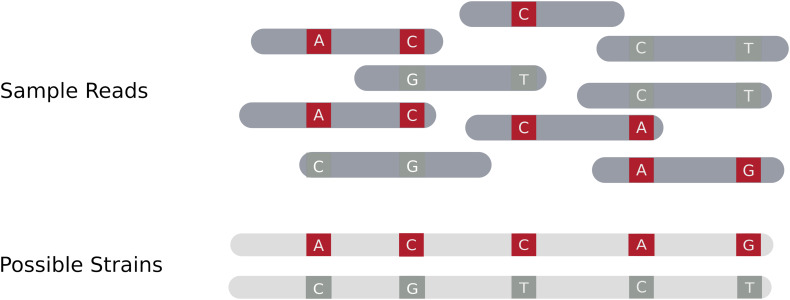
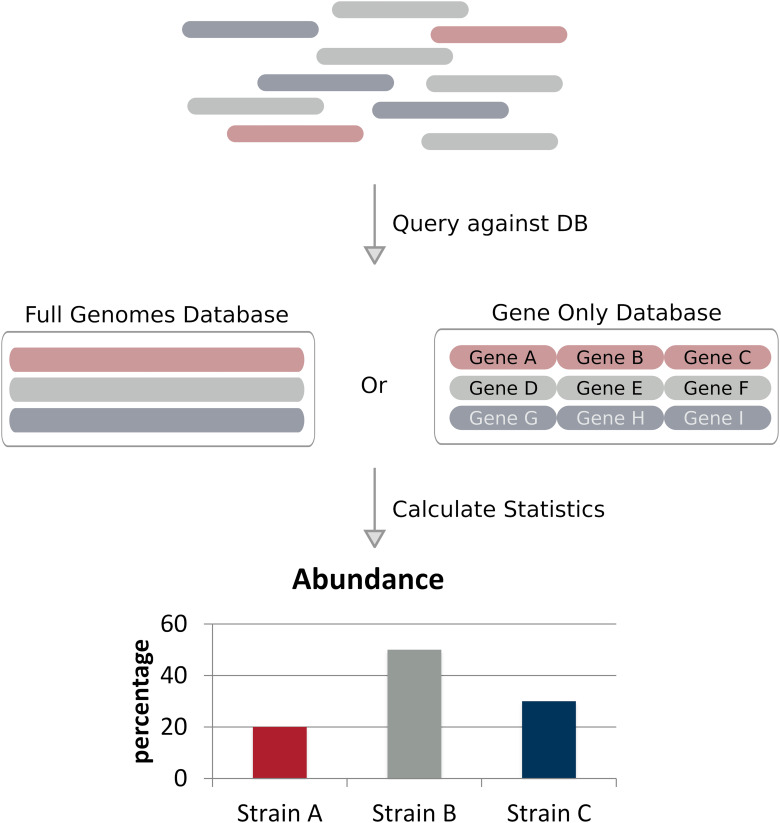
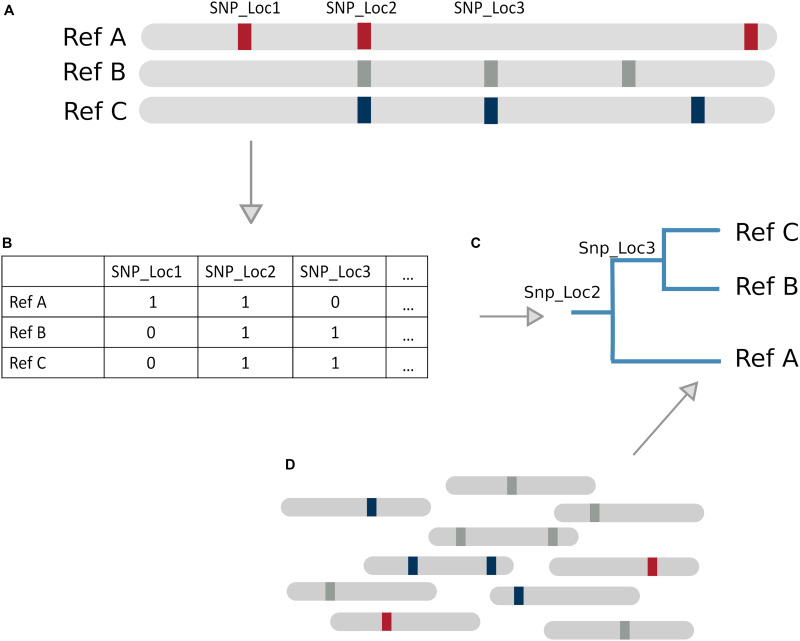
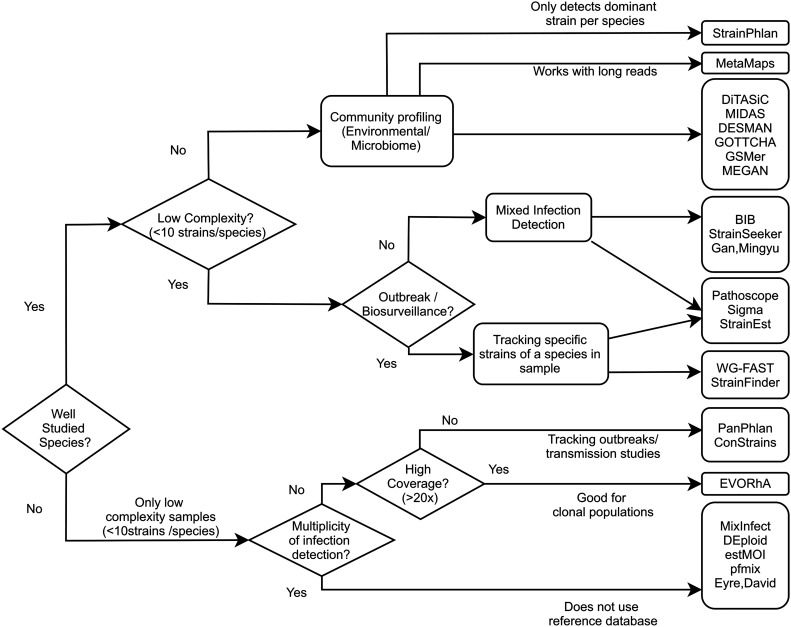
Metagenomic sequencing is a powerful tool for examining the diversity and complexity of microbial communities. Most widely used tools for taxonomic profiling of metagenomic sequence data allow for a species-level overview of the composition. However, individual strains within a species can differ greatly in key genotypic and phenotypic characteristics, such as drug resistance, virulence and growth rate. Therefore, the ability to resolve microbial communities down to the level of individual strains within a species is critical to interpreting metagenomic data for clinical and environmental applications, where identifying a particular strain, or tracking a particular strain across a set of samples, can help aid in clinical diagnosis and treatment, or in characterizing yet unstudied strains across novel environmental locations. Recently published approaches have begun to tackle the problem of resolving strains within a particular species in metagenomic samples. In this review, we present an overview of these new algorithms and their uses, including methods based on assembly reconstruction and methods operating with or without a reference database. While existing metagenomic analysis methods show reasonable performance at the species and higher taxonomic levels, identifying closely related strains within a species presents a bigger challenge, due to the diversity of databases, genetic relatedness, and goals when conducting these analyses. Selection of which metagenomic tool to employ for a specific application should be performed on a case-by case basis as these tools have strengths and weaknesses that affect their performance on specific tasks. A comprehensive benchmark across different use case scenarios is vital to validate performance of these tools on microbial samples. Because strain-level metagenomic analysis is still in its infancy, development of more fine-grained, high-resolution algorithms will continue to be in demand for the future.
Keywords: bioinformatics; metagenomics; methods review; microbial detection; strain-level classification; whole genome sequencing.
Copyright © 2020 Anyansi, Straub, Manson, Earl and Abeel.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources