

Distinct Clinical Features and Novel Mutations in Taiwanese Patients With X-Linked Agammaglobulinemia
- PMID: 33013854
- PMCID: PMC7498534
- DOI: 10.3389/fimmu.2020.02001
Distinct Clinical Features and Novel Mutations in Taiwanese Patients With X-Linked Agammaglobulinemia
Abstract
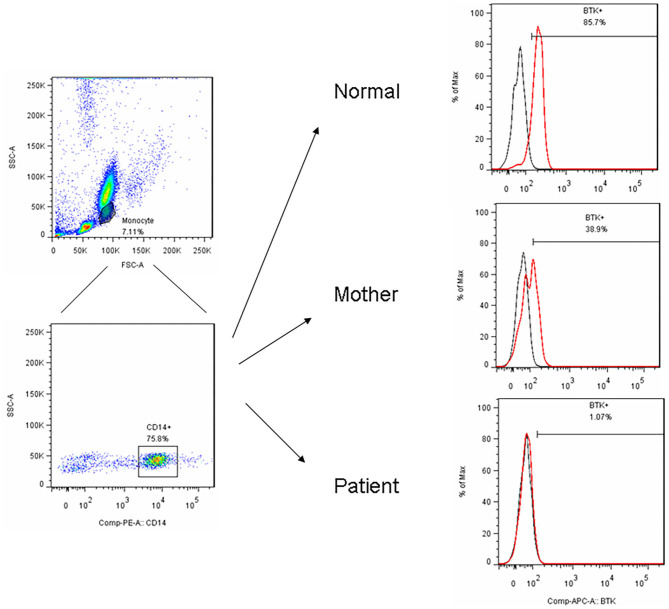
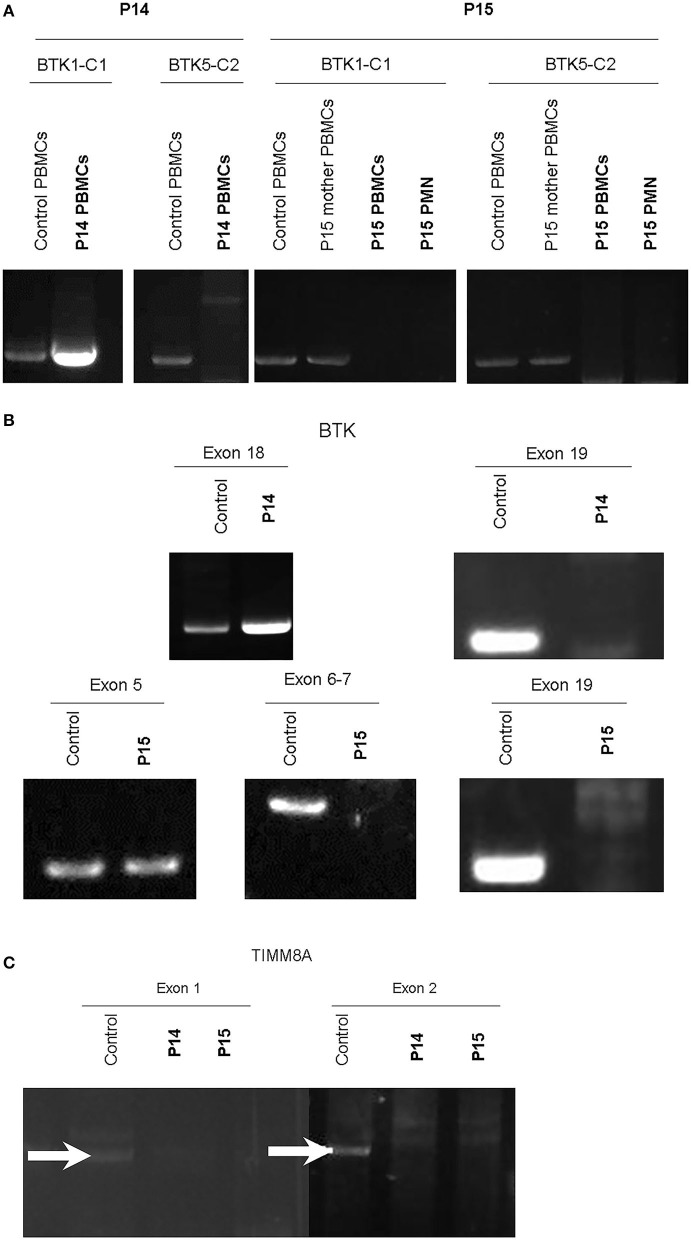
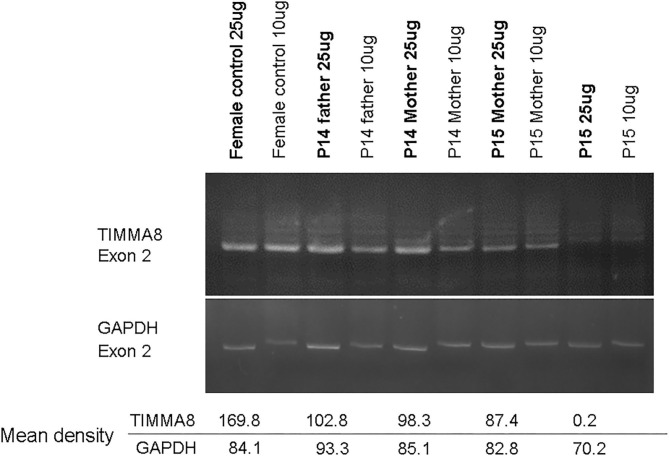
Background: X-linked agammaglobulinemia (XLA) is caused by a mutation of the Bruton's tyrosine kinase (BTK) gene and is the most common genetic mutation in patients with congenital agammaglobulinemia. The aim of this study was to analyze the clinical features, genetic defects, and/or BTK expression in patients suspected of having XLA who were referred from the Taiwan Foundation of Rare Disorders (TFRD). Methods: Patients with recurrent bacterial infections in the first 2 years of life, serum IgG/A/M below 2 standard deviations of the normal range, and ≦2% CD19+B cells were enrolled during the period of 2004-2019. The frequency of infections, pathogens, B-lymphocyte subsets, and family pedigree were recorded. Peripheral blood samples were sent to our institute for BTK expression and genetic analysis. Results: Nineteen (from 16 families) out of 29 patients had BTK mutations, including 7 missense mutations, 7 splicing mutations, 1 nonsense mutation, 2 huge deletions, and 2 nucleotide deletions. Six novel mutations were detected: c.504G>T [p.K168N], c.895-2A>G [p.Del K290 fs 23*], c.910T>G [p.F304V], c.1132T>C [p.T334H], c.1562A>T [p.D521V], and c.1957delG [Del p.D653 fs plus 45 a.a.]. All patients with BTK mutations had obviously decreased BTK expressions. Pseudomonas sepsis developed in 14 patients and led to both Shanghai fever and recurrent hemophagocytic lymphohistiocytosis (HLH). Recurrent sinopulmonary infections and bronchiectasis occurred in 11 patients. One patient died of pseudomonas sepsis and another died of hepatocellular carcinoma before receiving optimal treatment. Two patients with contiguous gene deletion syndrome (CGS) encompassing the TIMM8A/DDP1 gene presented with early-onset progressive post-lingual sensorineural Deafness, gradual Dystonia, and Optic Neuronopathy syndrome (DDON) or Mohr-Tranebjaerg syndrome (MTS). Conclusion: Pseudomonas sepsis was more common (74%) than recurrent sinopulmonary infections in Taiwanese XLA patients, and related to Shanghai fever and recurrent HLH, both of which were prevented by regular immunoglobulin infusions. Approximately 10% of patients belonged to CGS involving the TIMM8A/DDP1 gene and presented with the DDON/MTS phenotype in need of aggressive psychomotor therapy.
Keywords: Bruton's tyrosine kinase (BTK); Mohr-Tranebjaerg syndrome (MTS); TIMM8A/DDP1 gene; X-linked agammaglobulinemia (XLA); contiguous gene deletion syndrome (CGS); deafness-dystonia-optic neuronopathy syndrome (DDON).
Copyright © 2020 Yeh, Hsieh, Lee, Huang, Chen, Yeh, Ou, Yao, Wu and Lin.
Figures
References
-
- Bruton OC. Agammaglobulinemia. Pediatrics. (1952) 9:722–8. - PubMed
-
- Ohta Y, Haire RN, Litman RT, Fu SM, Nelson RP, Kratz J, et al. Genomic organization and structure of Bruton agammaglobulinemia tyrosine kinase: localization of mutations associated with varied clinical presentations and course in X chromosome-linked agammaglobulinemia. Proc Natl Acad Sci USA. (1994) 91:9062–6. 10.1073/pnas.91.19.9062 - DOI - PMC - PubMed
-
- Nomura K, Kanegane H, Karasuyama H, Tsukada S, Agematsu K, Murakami G, et al. Genetic defect in human X-linked agammaglobulinemia impedes amaturational evolution of pro-B cells into a later stage of pre-B cells in the B-cell differentiation pathway. Blood. (2000) 96:610–7. - PubMed
-
- Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine. (2006) 85:193–202. 10.1097/01.md.0000229482.27398.ad - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical