

doi: 10.1038/s41439-020-00114-w.
eCollection 2020.
Nonsense variants of STAG2 result in distinct congenital anomalies
Affiliations
- PMID: 33014403
- PMCID: PMC7501222
- DOI: 10.1038/s41439-020-00114-w
Item in Clipboard
Nonsense variants of STAG2 result in distinct congenital anomalies
Hum Genome Var.
.
Abstract
Herein, we report two female cases with novel nonsense mutations of STAG2 at Xq25, encoding stromal antigen 2, a component of the cohesion complex. Exome analysis identified c.3097 C>T, p.(Arg1033*) in Case 1 (a fetus with multiple congenital anomalies) and c.2229 G>A, p.(Trp743*) in Case 2 (a 7-year-old girl with white matter hypoplasia and cleft palate). X inactivation was highly skewed in both cases.
Keywords: Disease genetics; Next-generation sequencing.
© The Author(s) 2020.
Conflict of interest statement
Conflict of interestThe authors declare that they have no conflict of interest.
Figures
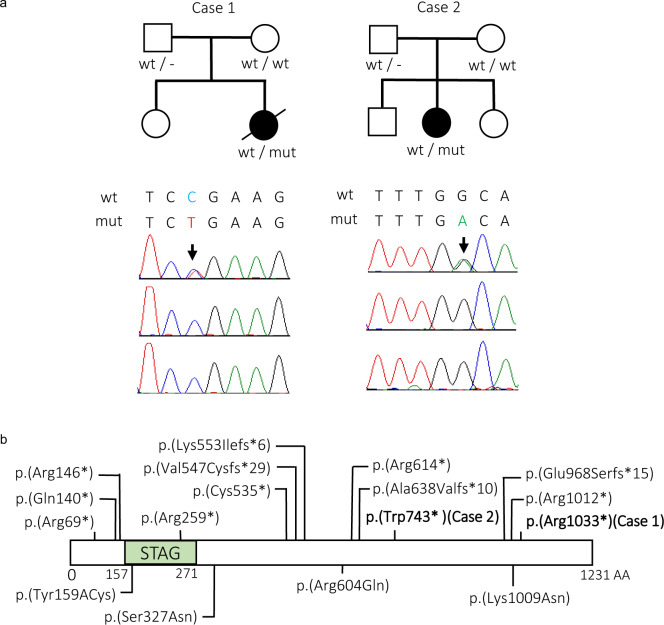
a Familial pedigrees and electropherograms of STAG2 variants [Case 1: c.3097 C>T: p.(Arg1033*), Case 2: c.2229 G>A, (p.Trp743*)]. The arrow indicates a heterozygous variant. wt, wild-type; mut, mutation. b Functional domain of the STAG2 protein and pathogenic variants. Truncating and missense variants are shown above and below the protein, respectively. Our cases are shown in bold. The STAG domain predicted by Pfam is shown (http://pfam.xfam.org ).

a The c.3097 C>T variant could be successfully mapped within the 450-kb phased haplotype block in Case 1 using HiFi sequence and haplotype phasing. b SNP typing confirmed that the mutation occurred in the paternal chromosome (Allele 2, the brown haplotype block in Fig. 2a). POS: position of sequence, Pt: patient, Fa: father, Mo: mother.
References
LinkOut - more resources
Full Text Sources
