

Relationships between Mitochondrial Dysfunction and Neurotransmission Failure in Alzheimer's Disease
- PMID: 33014538
- PMCID: PMC7505271
- DOI: 10.14336/AD.2019.1125
Relationships between Mitochondrial Dysfunction and Neurotransmission Failure in Alzheimer's Disease
Abstract
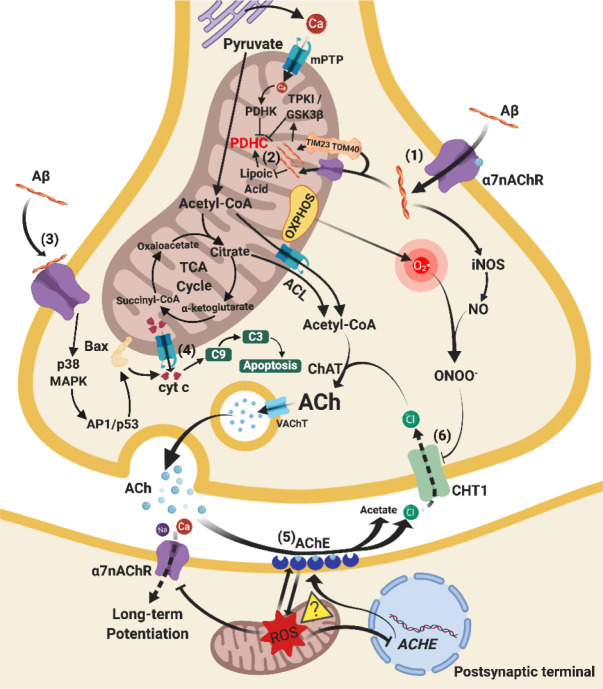
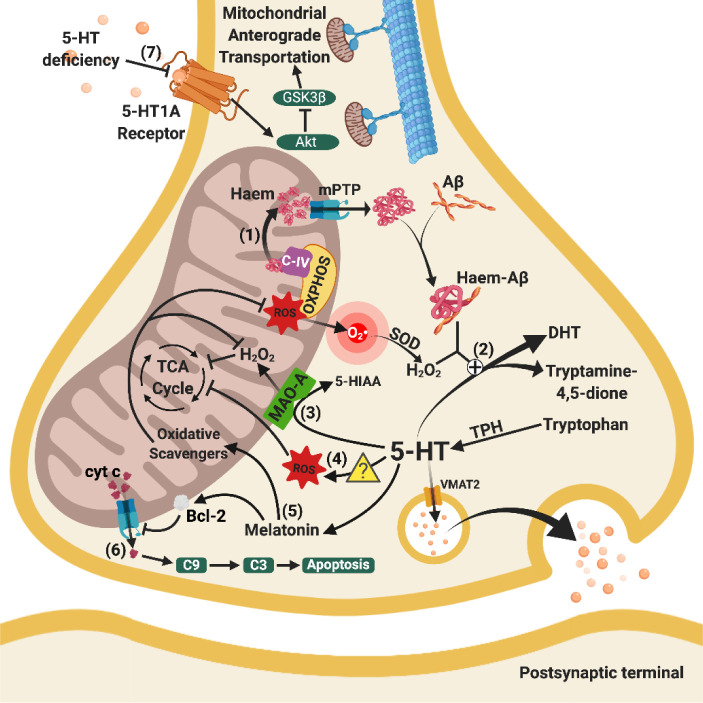
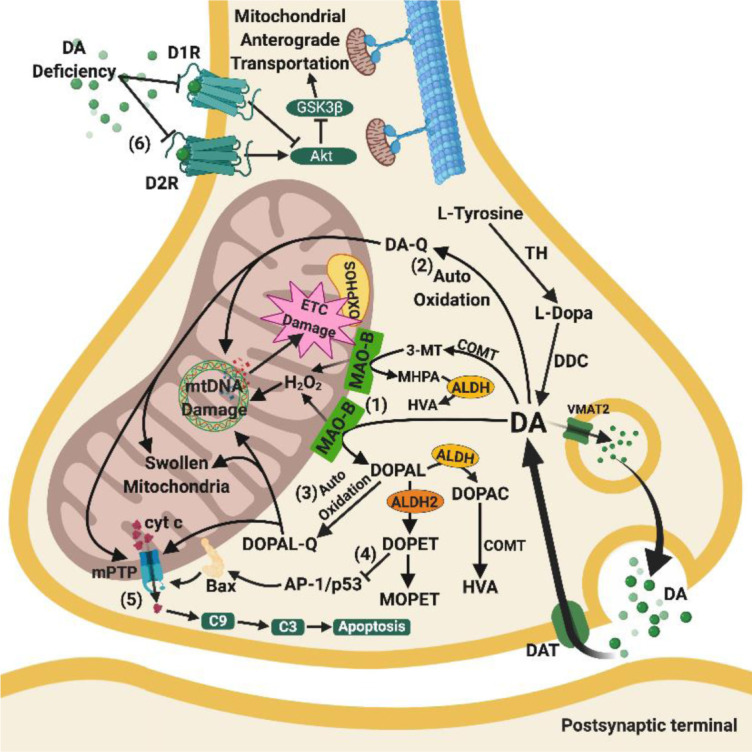
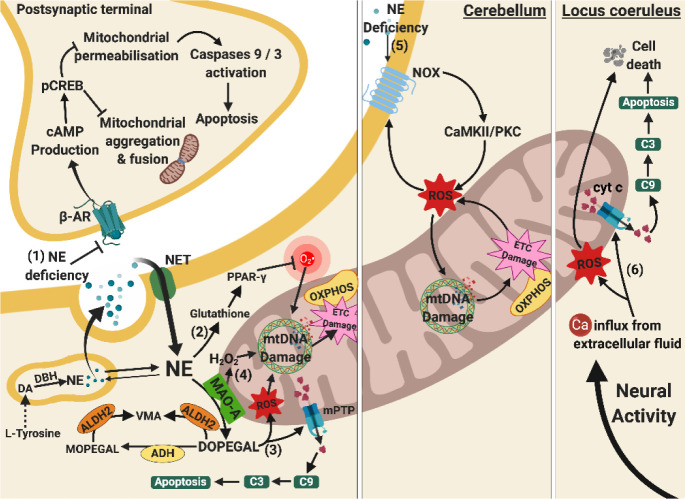
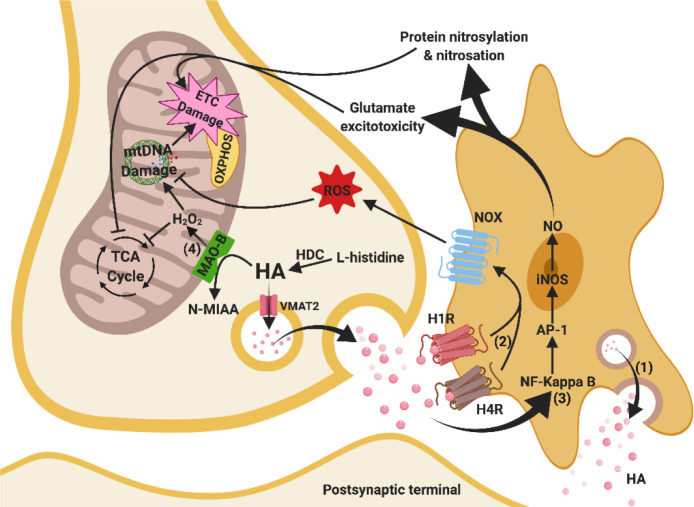
Besides extracellular deposition of amyloid beta and formation of phosphorylated tau in the brains of patients with Alzheimer's disease (AD), the pathogenesis of AD is also thought to involve mitochondrial dysfunctions and altered neurotransmission systems. However, none of these components can describe the diverse cognitive, behavioural, and psychiatric symptoms of AD without the pathologies interacting with one another. The purpose of this review is to understand the relationships between mitochondrial and neurotransmission dysfunctions in terms of (1) how mitochondrial alterations affect cholinergic and monoaminergic systems via disruption of energy metabolism, oxidative stress, and apoptosis; and (2) how different neurotransmission systems drive mitochondrial dysfunction via increasing amyloid beta internalisation, oxidative stress, disruption of mitochondrial permeabilisation, and mitochondrial trafficking. All these interactions are separately discussed in terms of neurotransmission systems. The association of mitochondrial dysfunctions with alterations in dopamine, norepinephrine, and histamine is the prospective goal in this research field. By unfolding the complex interactions surrounding mitochondrial dysfunction in AD, we can better develop potential treatments to delay, prevent, or cure this devastating disease.
Keywords: Alzheimer’s disease; mitochondrial dysfunction; monoaminergic; neurotransmission dysfunction.
copyright: © 2020 Wong et al.
Conflict of interest statement
Declaration of Competing Interest None.
Figures
References
-
- Alzheimer’s Association (2018). 2018 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 14:367-429.
-
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP (2013). The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s & Dementia, 9:63-75.e62. - PubMed
-
- Hardy JA, Higgins GA (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science, 256:184-185. - PubMed
-
- Querfurth HW, LaFerla FM (2010). Alzheimer’s disease. N Engl J Med, 362:329-344. - PubMed
Publication types
LinkOut - more resources
Full Text Sources