

Sickle cell disease in the era of precision medicine: looking to the future
- PMID: 33015364
- PMCID: PMC7531762
- DOI: 10.1080/23808993.2019.1688658
Sickle cell disease in the era of precision medicine: looking to the future
Abstract
Introduction: Sickle cell anemia is a mendelian disease that is noted for the heterogeneity of its clinical expression. Because of this, providing an accurate prognosis has been a longtime quest.
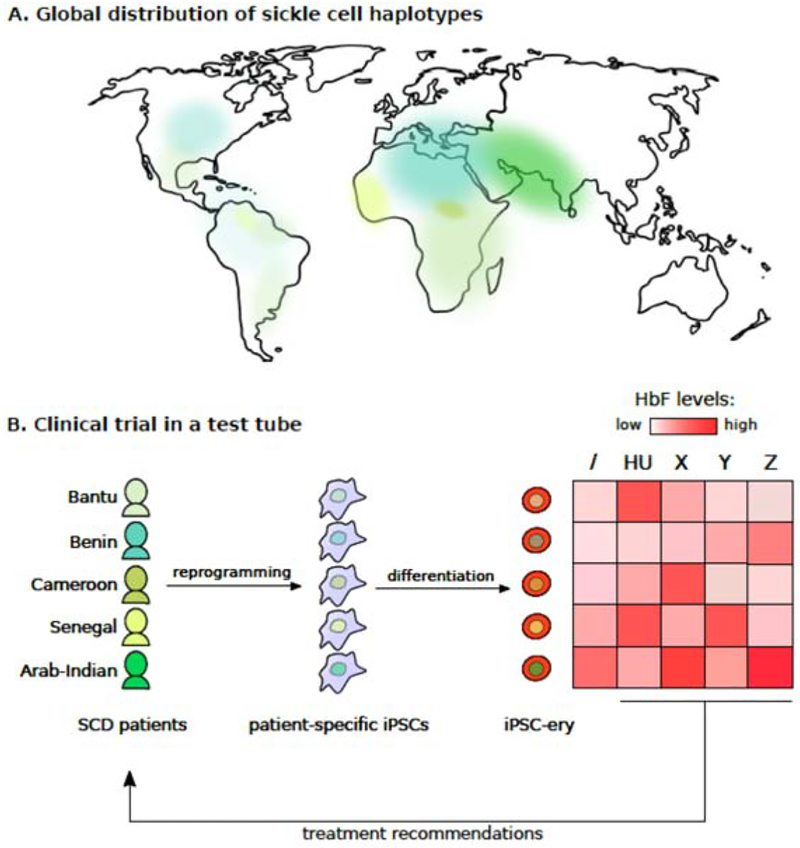
Areas covered: Reviewed are the benefits and shortcomings of testing for the major modulators of the severity of disease, like fetal hemoglobin and α thalassemia, along with studies that have attempted to link genetic variation with sub-phenotypes of disease in a predictive fashion. Induced pluripotent stem cells driven to differentiate into erythroid precursor cells provide another area for potential patient-specific drug testing.
Expert opinion: Fetal hemoglobin is the strongest modulator of sickle cell anemia but simply measuring its blood levels is an insufficient means of forecasting an individual's prognosis. A more precise method would be to know the distribution of fetal hemoglobin levels across the population of red cells, an assay not yet available. Prognostic measures have been developed using genetic and other signatures, but their predictive value is suboptimal. Widely applicable assays must be developed to allow a tailored approach to using the several new treatments that are likely to be available in the near future.
Keywords: Fetal hemoglobin; GWAS; SNPs; genetic signatures; hemolysis; iPSCs; vasoocclusion.
Conflict of interest statement
Declaration of interest The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Figures
References
-
- Garner C, Tatu T, Reittie JE, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood 2000;95:342–346. - PubMed
-
- Dover GJ, Boyer SH, Charache S, et al. Individual Variation in the Production and Survival of F Cells in Sickle-Cell Disease. N. Engl. J. Med 1978;299:1428–1435. - PubMed
-
- Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Invest 2017;127:750–760. - PMC - PubMed
-
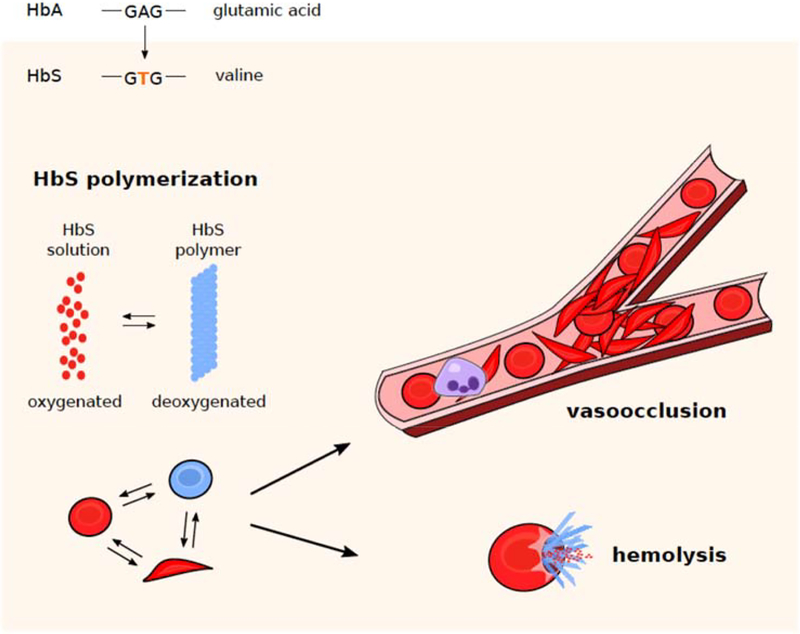
This review provides background and evidence for the dichotomization of sickle cell pathophysiology into events associated with vasoocclusion and intravascular hemolysis.
Grants and funding
LinkOut - more resources
Full Text Sources