

DDX5 resolves R-loops at DNA double-strand breaks to promote DNA repair and avoid chromosomal deletions
- PMID: 33015627
- PMCID: PMC7520851
- DOI: 10.1093/narcan/zcaa028
DDX5 resolves R-loops at DNA double-strand breaks to promote DNA repair and avoid chromosomal deletions
Abstract
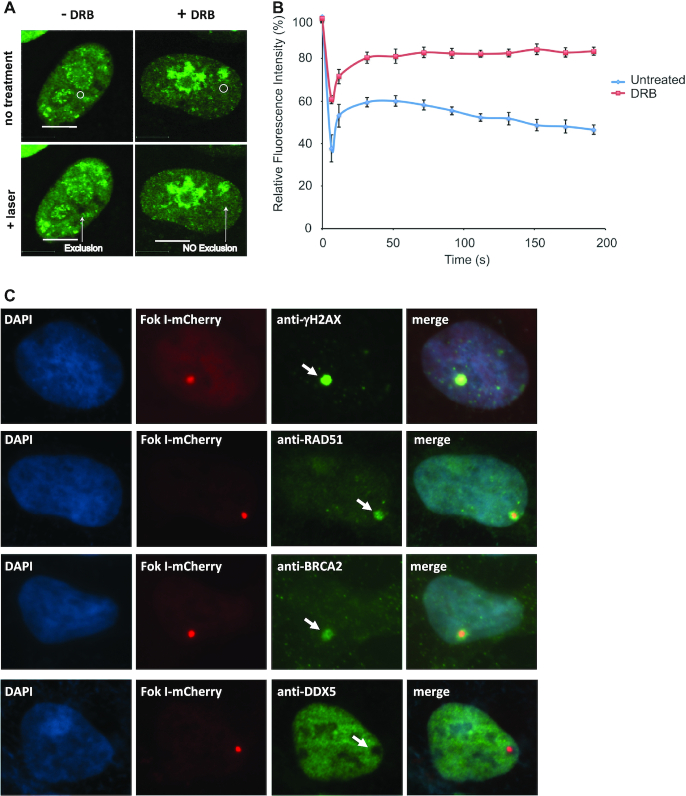
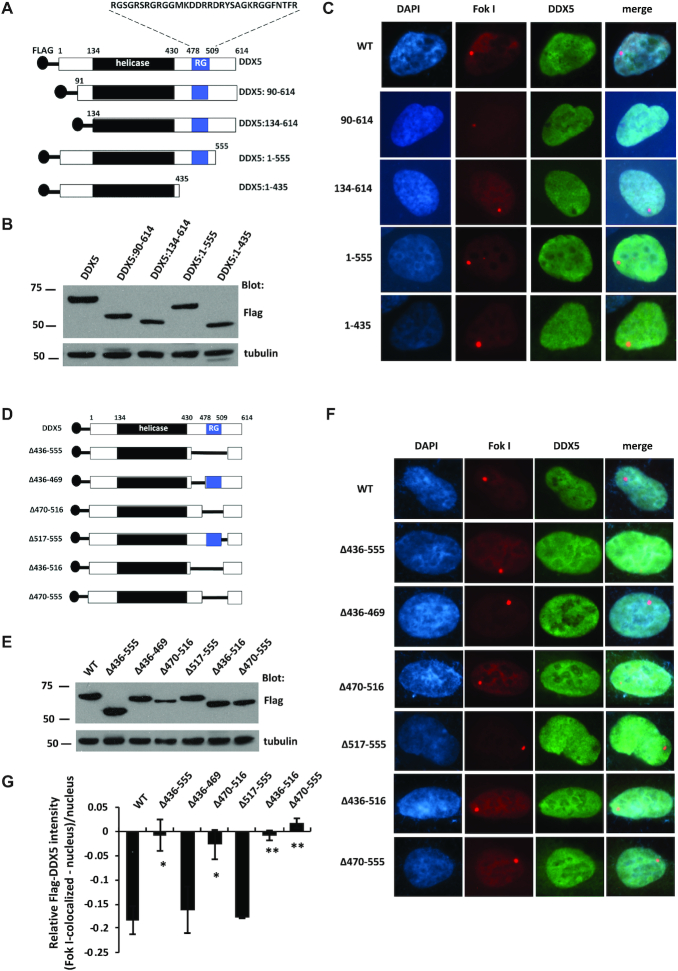
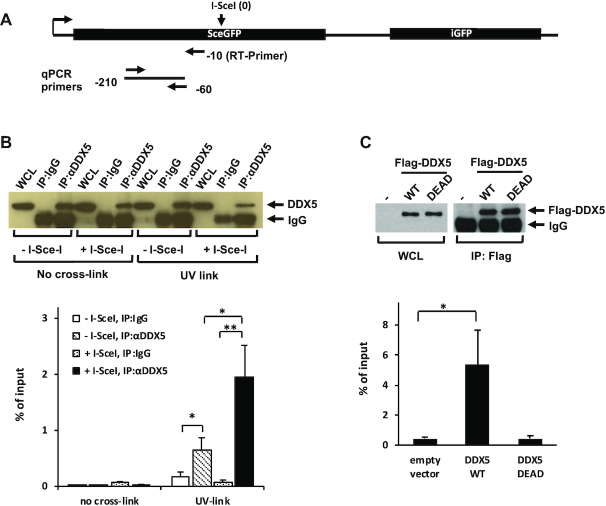
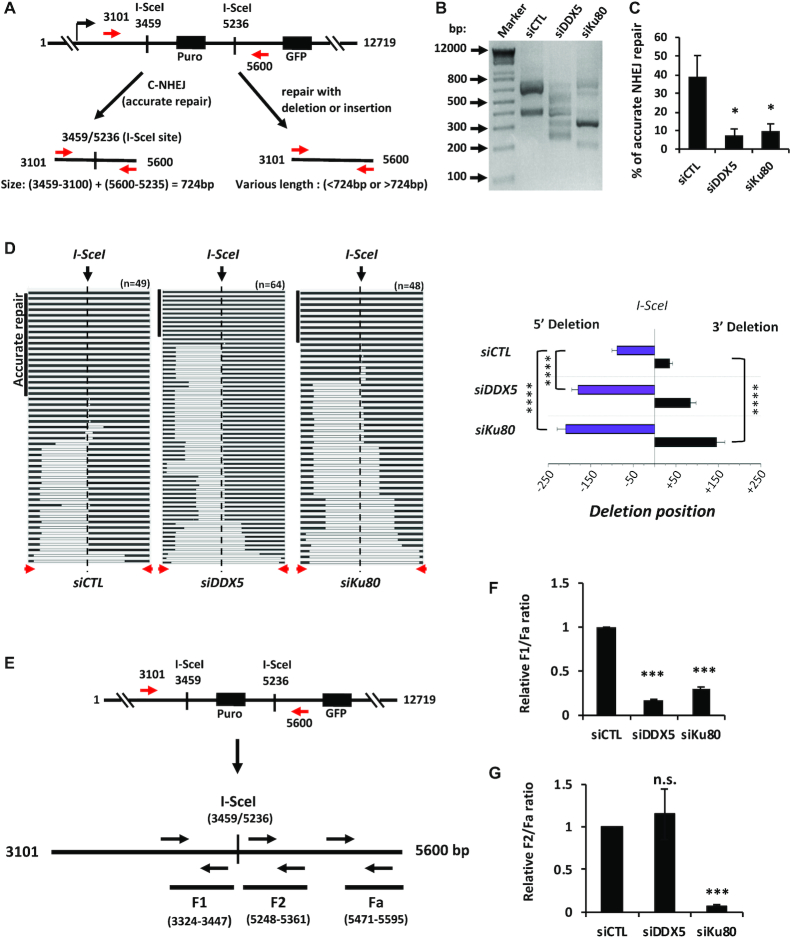
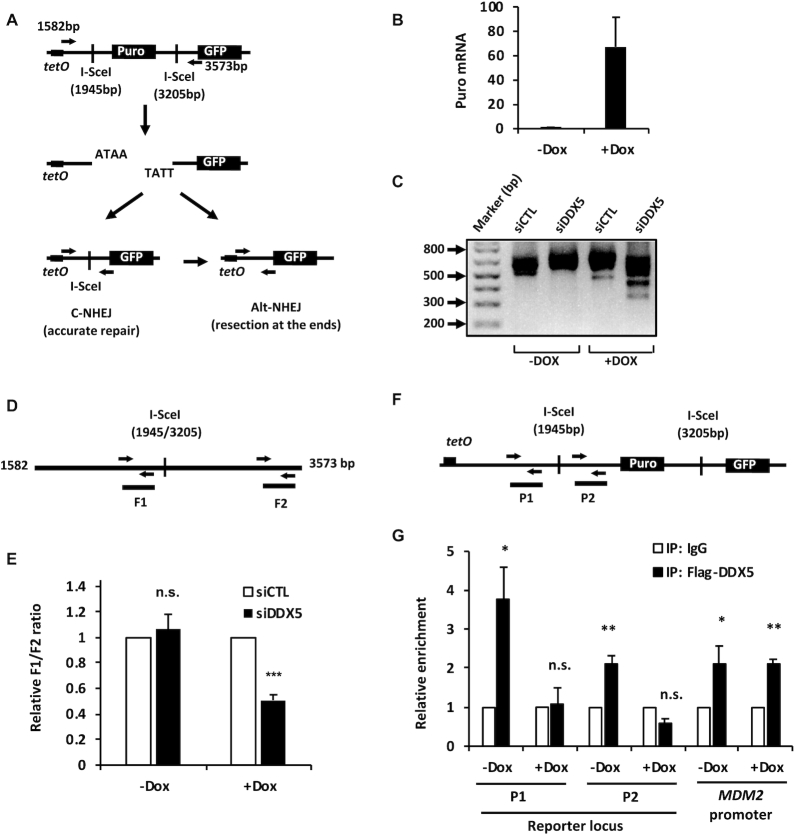
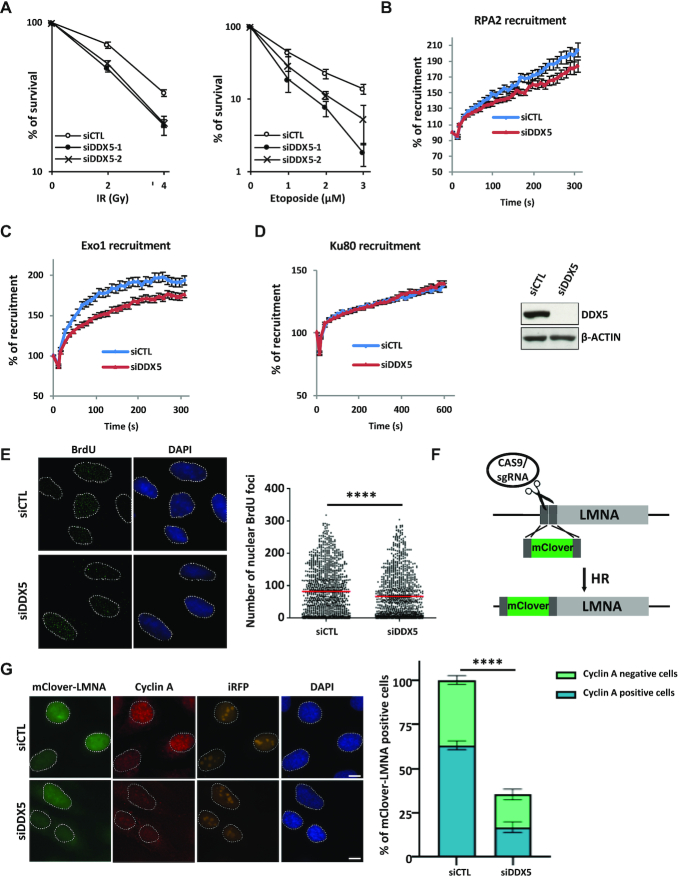
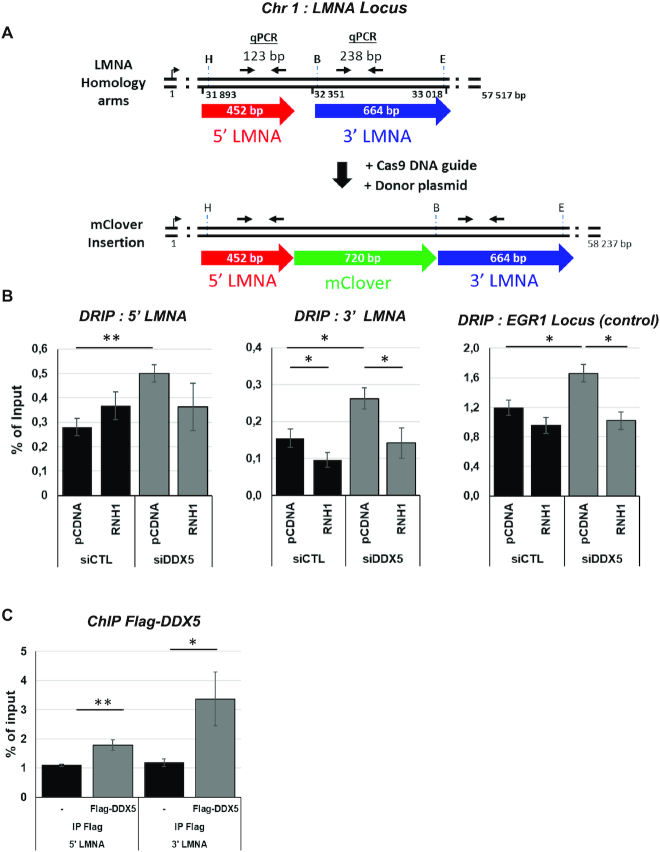
R-loops are three-stranded structures consisting of a DNA/RNA hybrid and a displaced DNA strand. The regulatory factors required to process this fundamental genetic structure near double-strand DNA breaks (DSBs) are not well understood. We previously reported that cellular depletion of the ATP-dependent DEAD box RNA helicase DDX5 increases R-loops genome-wide causing genomic instability. In this study, we define a pivotal role for DDX5 in clearing R-loops at or near DSBs enabling proper DNA repair to avoid aberrations such as chromosomal deletions. Remarkably, using the non-homologous end joining reporter gene (EJ5-GFP), we show that DDX5-deficient U2OS cells exhibited asymmetric end deletions on the side of the DSBs where there is overlap with a transcribed gene. Cross-linking and immunoprecipitation showed that DDX5 bound RNA transcripts near DSBs and required its helicase domain and the presence of DDX5 near DSBs was also shown by chromatin immunoprecipitation. DDX5 was excluded from DSBs in a transcription- and ATM activation-dependent manner. Using DNA/RNA immunoprecipitation, we show DDX5-deficient cells had increased R-loops near DSBs. Finally, DDX5 deficiency led to delayed exonuclease 1 and replication protein A recruitment to laser irradiation-induced DNA damage sites, resulting in homologous recombination repair defects. Our findings define a role for DDX5 in facilitating the clearance of RNA transcripts overlapping DSBs to ensure proper DNA repair.
© The Author(s) 2020. Published by Oxford University Press on behalf of NAR Cancer.
Figures

References
-
- García-Muse T., Aguilera A. R loops: from physiological to pathological roles. Cell. 2019; 179:604–618. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
