

Epigenetic modulator inhibition overcomes temozolomide chemoresistance and antagonizes tumor recurrence of glioblastoma
- PMID: 33016927
- PMCID: PMC7598052
- DOI: 10.1172/JCI127916
Epigenetic modulator inhibition overcomes temozolomide chemoresistance and antagonizes tumor recurrence of glioblastoma
Abstract
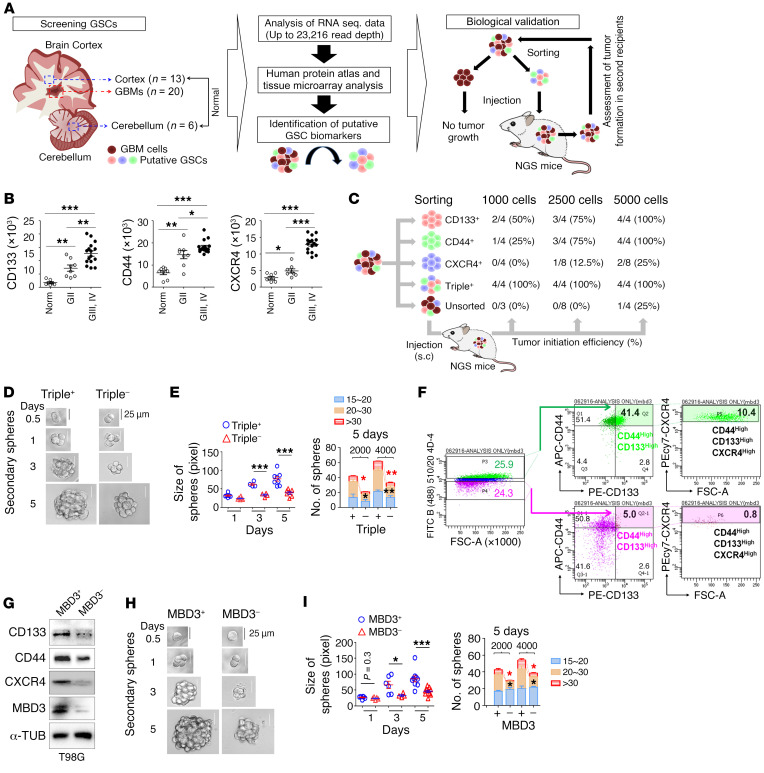
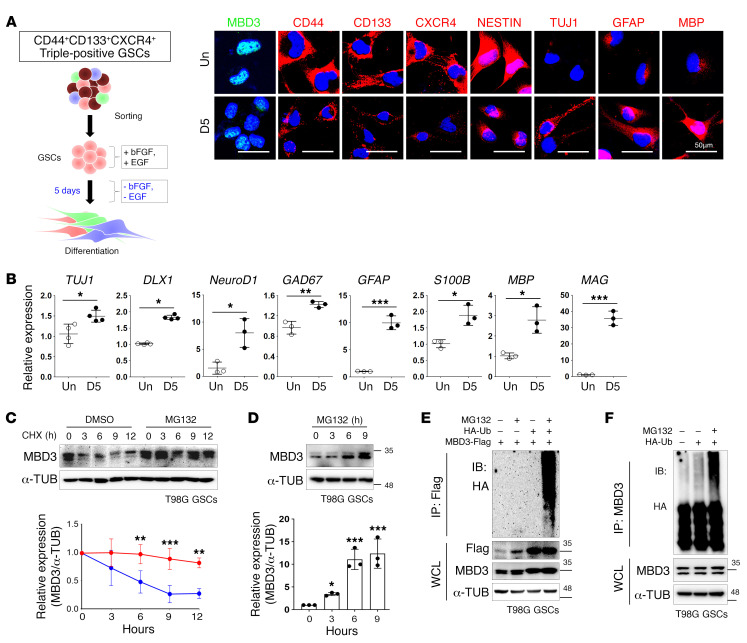
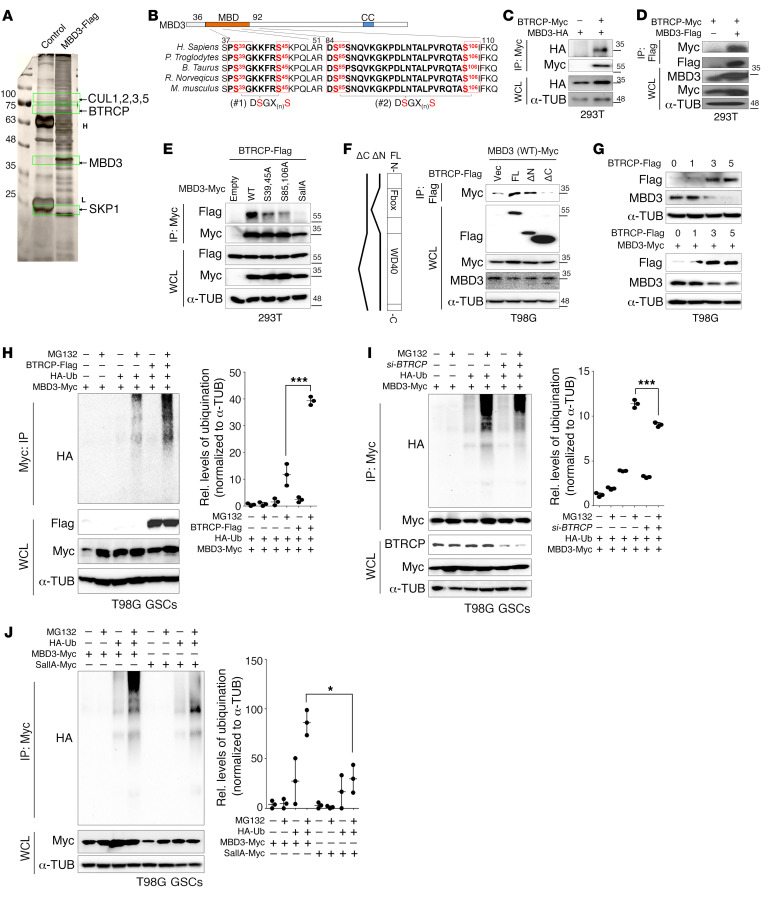
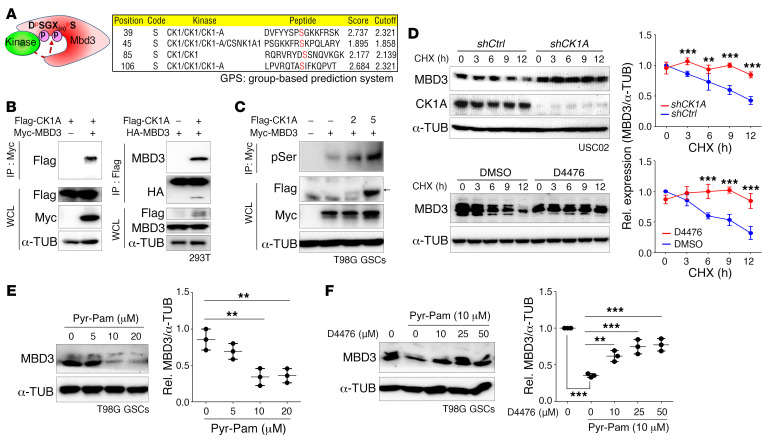
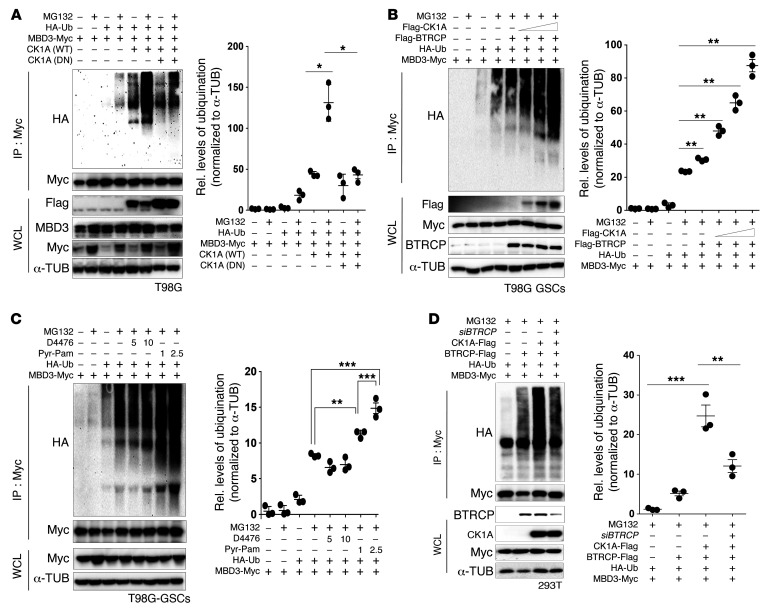
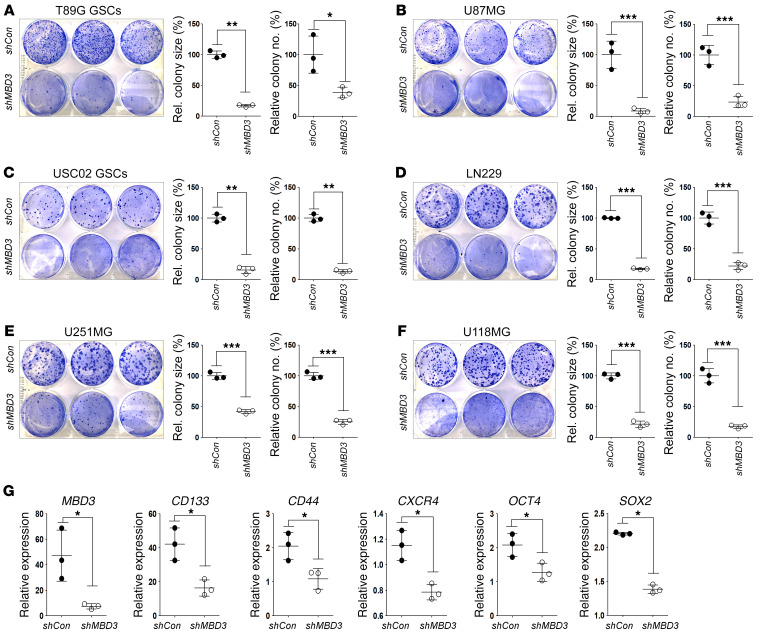
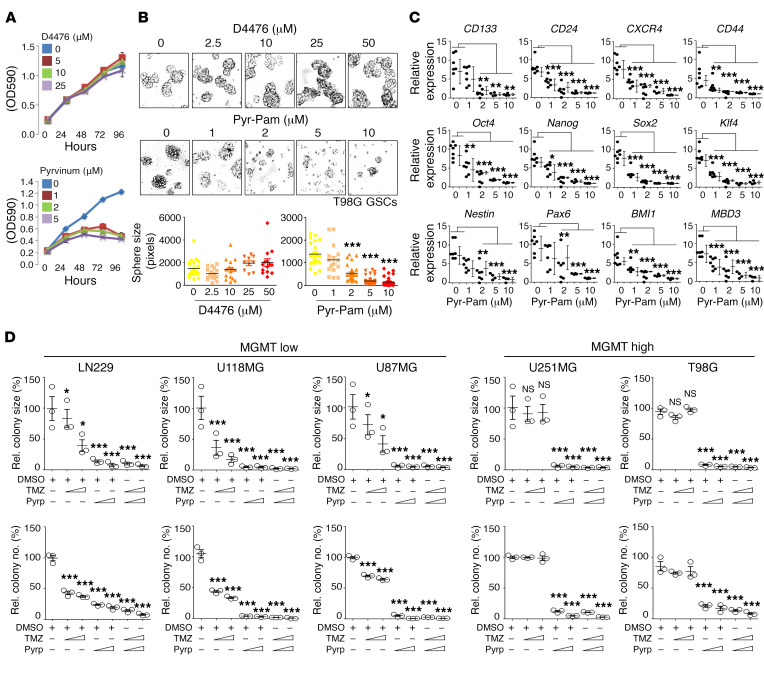
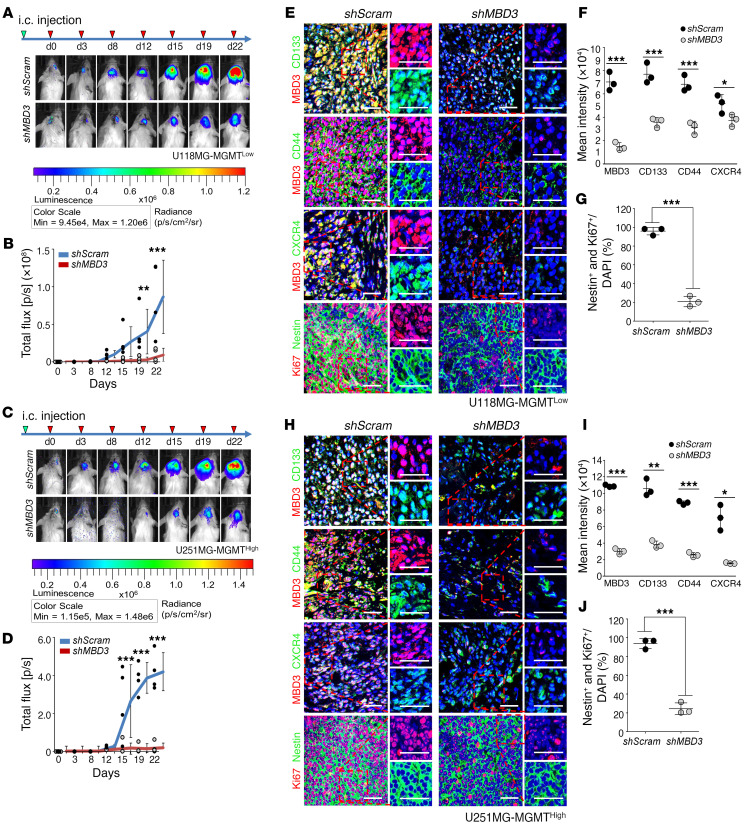
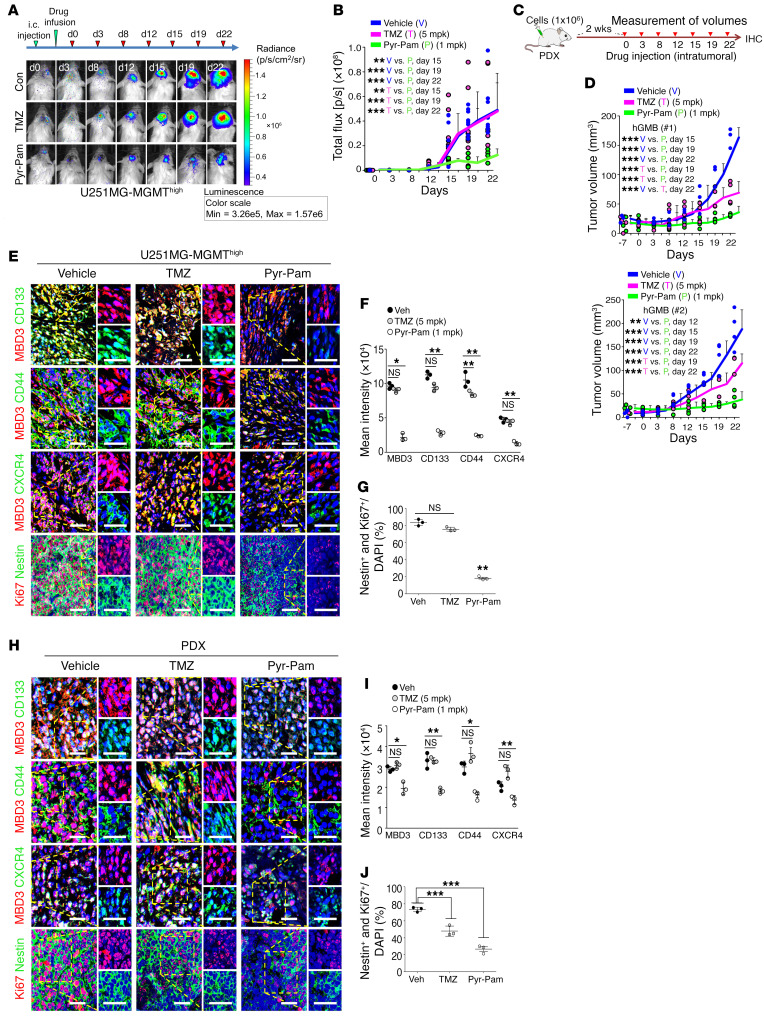
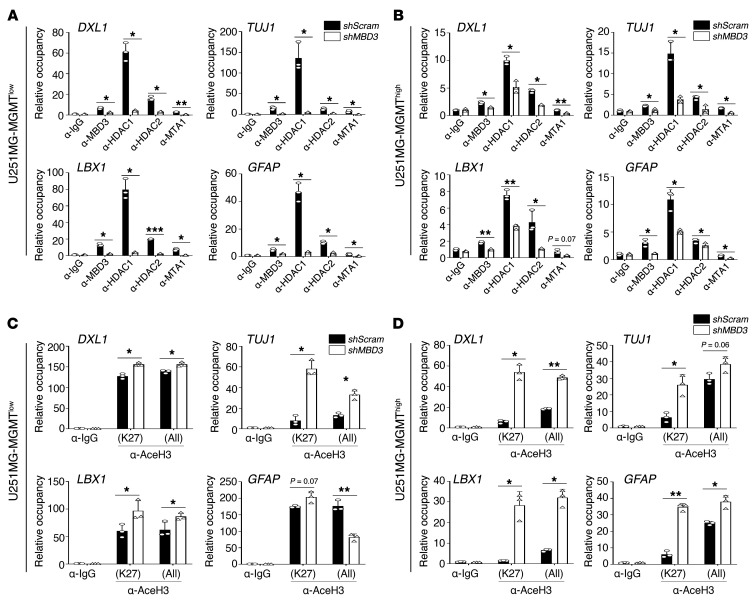
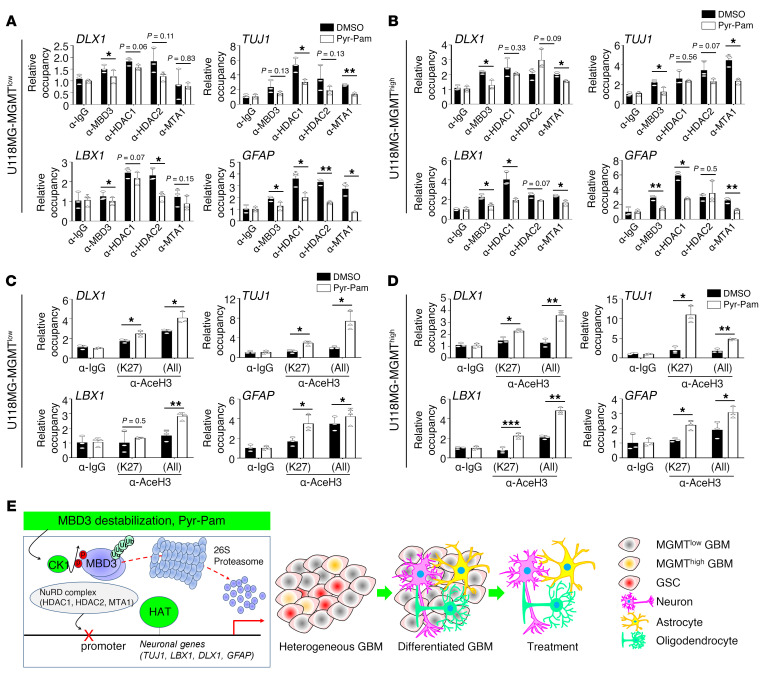
Glioblastoma multiforme (GBM) heterogeneity causes a greater number of deaths than any other brain tumor, despite the availability of alkylating chemotherapy. GBM stem-like cells (GSCs) contribute to GBM complexity and chemoresistance, but it remains challenging to identify and target GSCs or factors that control their activity. Here, we identified a specific GSC subset and show that activity of these cells is positively regulated by stabilization of methyl CpG binding domain 3 (MBD3) protein. MBD3 binds to CK1A and to BTRCP E3 ubiquitin ligase, triggering MBD3 degradation, suggesting that modulating this circuit could antagonize GBM recurrence. Accordingly, xenograft mice treated with the CK1A activator pyrvinium pamoate (Pyr-Pam) showed enhanced MBD3 degradation in cells expressing high levels of O6-methylguanine-DNA methyltransferase (MGMT) and in GSCs, overcoming temozolomide chemoresistance. Pyr-Pam blocked recruitment of MBD3 and the repressive nucleosome remodeling and deacetylase (NuRD) complex to neurogenesis-associated gene loci and increased acetyl-histone H3 activity and GSC differentiation. We conclude that CK1A/BTRCP/MBD3/NuRD signaling modulates GSC activation and malignancy, and that targeting this signaling could suppress GSC proliferation and GBM recurrence.
Keywords: Brain cancer; Drug therapy; Epigenetics; Oncology; Stem cells.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
